


























































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Cancer Grand Rounds: HER2 und EGFR: Molekulare Mechanismen und Targets für die nächste Generation von Tumortherapien
24. Februar 2012
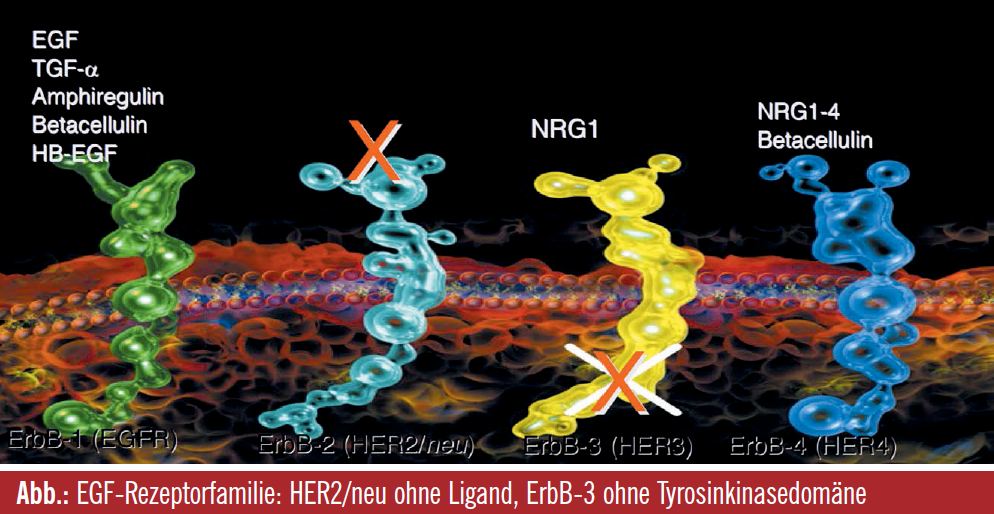
Erlotinib, Gefitinib, Lapatinib, Cetuximab, Panitumumab, Pertuzumab und Trastuzumab sind Beispiele für Substanzen, die, obwohl sie alle gegen eine Rezeptorfamilie gerichtet sind, bei unterschiedlichen Tumoren zur Anwendung kommen (Brust, Lunge, Kopf-Hals-Bereich, Magen, Pankreas), woran ersichtlich wird, dass grundlegende Prozesse der Tumorentstehung und Metastasierung von dieser Rezeptorfamilie kontrolliert werden. Als jungem Wissenschaftler waren für Yosef Yarden zwei Beobachtungen interessant: Die Feststellung, dass HER2 mit seiner im humanen Kinom möglicherweise stärksten onkogenen Kinase keine Liganden besitzt, d. h. es können keine intrinsischen Wachstumsfaktoren gebunden werden und – Hand in Hand damit – dass der HER3- Rezeptor keine Tyrosinkinasedomäne besitzt und damit per se keine Signale weiterleiten kann, selbst wenn Liganden für diesen Rezeptor vorhanden sind (so als wäre genügend Treibstoff für ein Auto vorhanden, das aber keinen Motor hat). Offenbar war es in der Evolution wesentlich, einen Rezeptor ohne Liganden und einen ohne Tyrosinkinasedomäne beizubehalten. In jungen Jahren, meinte Yosef Yarden, wäre er davon überzeugt gewesen, dass man bei genauer Suche auch einen Wachstumsfaktor für HER2 finden müsste, was zwar nicht gelungen ist, aber die Entdeckung des Liganden Neuregulin-4, der spezifisch nur an ErbB4 bindet, geht auf Yarden zurück.
Genomduplikationen und Subfunktionalisierung: Die Komplexität des EGFRNetzwerks wurde als Resultat von Genomduplikationen beschrieben. Ausgehend von nur einem Rezeptor, der im „Modellorganismus“ C. elegans in linearer Weise mit nur einem EGF-Molekül interagieren konnte, bildeten sich in jeweils höher entwickelten Spezies bis hin zum Menschen 2, 3 und 4 Rezeptoren aus, die mit der Möglichkeit zur Homo- und Heterodimerisierung und mit mehr Liganden ausgestattet waren. So wurde ein Netzwerk kreiert, das durch Diversität und Subfunktionalisierung (dem Zugewinn neuer Eigenschaften) immer robuster gegenüber äußeren Einflüssen wurde und Störungen durch alternative Umgehungswege kompensieren konnte („Plastizität“ des Netzwerks). Als Produkt der Subfunktionalisierung wurde beispielsweise die Bildung von HER2- und HER3-assoziierten Heterodimeren beschrieben, die mit dem MAP- oder AKT-Kinase-Pathway interagieren und auf diese Weise hoch mitogen sind. Die Diversität der EGF-Rezeptor-Familie zeigt sich u. a. an der Beobachtung, dass bei verschiedenen Tumoren jeweils andere Charakteristika in den Vordergrund treten: So finden sich Deletionen und konstitutiv aktivierte EGF-Rezeptoren bei malignen Hirntumoren oder Punktmutationen beim NSCLC, die prädiktiv für das Ansprechen auf Tyrosinkinasehemmer sind. ErbB4 liegt z. B. beim malignen Melanom mutiert vor. Demgegenüber zeigt HER2 nur ganz selten Mutationen, wird aber aufgrund der Genamplifikation von Chromosom 17 beim Mammakar zinom überexprimiert. Neben mutationsreichen Varianten und der Genamplifikation wurde auch die autokrine Rezeptorstimulation als „Hallmark“ hervorgehoben, mit der Tumorzellen die zur Proliferation benötigten Wachstumsfaktoren selbst produzieren.
Deregulierte Endozytose als „Hallmark“ der Onkologie: Dem HER2-Molekül, dem in der Evolution ein Ligand verweigert wurde, kommt in diesem Netzwerk eine Schlüsselrolle beispielsweise als Feedback-Kontrolle zu. Während ein physiologischer Vorgang darin besteht, dass Rezeptoren nach ihrer Stimulation internalisiert und lysosomal abgebaut werden, wird dieser Prozess durch das HER2-Molekül verhindert, das den EGF-Rezeptor im Endosom recycelt, worauf wieder ein intaktes Molekül an der Zelloberfläche exprimiert werden kann. HER2 manipuliert auf diese Weise die negative Feedback-Kontrolle und hält die Proliferationskapazität durch positive Feedback-Schleifen stabil. Hier könnten HER2-degradierende Hitzeschockprotein-Inhibitoren interessant sein.
„2-Hit-Theorie“ zur Entstehung invasiver Mammakarzinome: Ein interessantes Beispiel für die Ausbildung eines HER2-assoziierten malignen Phänotyps ist die Beobachtung, dass in normalen Milchgängen der Brustdrüse kein HER2-überexprimiert wird, während HER2 beim duktalen Carcinoma in situ und beim intraduktalen Karzinom zu gewissen Anteilen nachweisbar ist. In diesem Kontext wurde gezeigt, dass die HER2- Überexpression (1. „Hit“) die intraluminale Apoptose von Brustkrebszellen verhindert und dass unter Zugabe von Wachstumsfaktoren (2. „Hit“) mit Hochregulation von TGF-β die extrazelluläre Matrix degradiert wird und ein invasiver Phänotyp entsteht („2-Hit-Theorie“ zur Entstehung invasiver Mammakarzinome). Frühe antiapoptotische Charakteristika gehen auf die Aktivierung des Notch-Pathways zurück, wie in Kooperation der Medizinischen Universität Wien mit dem Weizmann Institut von Prof. Wolfgang Köstler untersucht wurde.
Switch on, switch off: Sobald mitogene Signale an den Zellkern gelangt sind, werden Transkriptionsfaktoren (wie c- Fos, c-Jun, c-Myc) aktiviert, die als Protoonkogene bestimmte Genabschnitte regulieren. Normalerweise werden diese Gene nach der ersten Welle der Aktivierung (early genes, „switch on“) durch Transkriptionsfaktor-Repressoren (z. B. RNA-Bindungsproteine) im Sinne einer negativen Feedbackkontrolle wieder abgeschaltet (delayed early genes, „switch off“). Dieser Feedback-Mechanismus ist bei Tumoren ebenfalls herunterreguliert, während transkriptionelle „Switch on“-Faktoren hochreguliert sind. In dem Zusammenhang wurde auch untersucht, unter welchen Voraussetzungen eine Zelle in den Zellzyklus eintritt. Offenbar braucht es dafür entweder eine länger dauernde kontinuierliche Stimulation mit Wachstumsfaktoren oder aber die Stimulation durch zumindest zwei Wachstumsfaktor-Pulse, andernfalls passieren Zellen nicht den kritischen Punkt in der G1-Phase, an dem sich entscheidet, ob der Rest des Zellzyklus durchlaufen wird. Eine Hypothese besteht darin, dass der erste Puls zugleich auch p53-assoziierte Suppressorgene aktiviert, die den Eintritt in den Zellzyklus verhindern, während der 2. Puls die Assoziation mit p53 auflöst. p53 wirkt in diesem Modell als Filter vor überschießender Stimulation durch ein Wachstumsfaktor-„Hintergrundrauschen“. Dieser Filter ist bei Tumorzellen ebenfalls defekt. Laut Yarden reicht neben der mit Überexpression einhergehenden kontinuierlichen Stimulation bereits ein Puls, um den p53-assoziierten Filter auszuschalten (z. B. über den PIK3-Kinase-Pathway) und die Tumorzelle zur Proliferation anzuregen.
Ungewöhnliche, „trickreiche“ Angriffe: Im Kontext der Tumorbiologie zielt das EGFR-HER2-Programm auf Proliferation und Metastasierung. Wenn Netzwerke – funktionsregulierende Elemente aus DNA, RNA, Proteinen und kleinen Molekülen – expandieren, steigen die Interaktionsmöglichkeiten zwischen den Schaltstellen mit dem Ziel, gegenüber Störfaktoren stabil zu bleiben. All das wäre frustran, gäbe es nicht als kritischen Punkt die Fragilität dieser Netzwerke gegenüber ungewöhnlichen, d. h. in der Evolution nicht stattgefundenen, z. B. von mehreren Seiten gleichzeitig erfolgenden Angriffen auf einen zentralen Knotenpunkt (es werden mehr Züge ausfallen, wenn der Zentralbahnhof betroffen ist, als wenn ein Nebengleis betroffen ist) – wobei es prinzipiell einleuchtet, dass Kombinationstherapien mehrere schaltstellenabhängige Signalwege destabilisieren und das Netzwerk auf diese Weise eher kollabiert oder Resistenzen später auftreten. Ein Ansatz von Yosef Yarden, der dahingehend auch einige Entwicklungen patentiert hat, sind Antikörperkombinationen mit synergistisch wirkenden epitopspezifischen Angriffspunkten – wobei die Antikörpertherapie, passiv oder aktiv, auch insofern interessant ist, als bislang keine Autoantikörper gegen EGFR oder HER2 gefunden wurden, womit die Rekrutierung des Immunsystems eine jener „trickreichen“ Facetten ist, die das Netzwerk zu Fall bringen können.

Ursprünglich erschienen:
SO 01|2012
SO 01|2012
Herausgeber: Univ.-Prof. Dr. Christoph Zielinksi, Univ.-Prof. Dr. Markus Raderer
Publikationsdatum: 2012-02-24
Zur Ausgabe »
Publikationsdatum: 2012-02-24
Zur Ausgabe »