


























































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Individuelle Dosisanpassung in der Tumortherapie
23. Dezember 2011
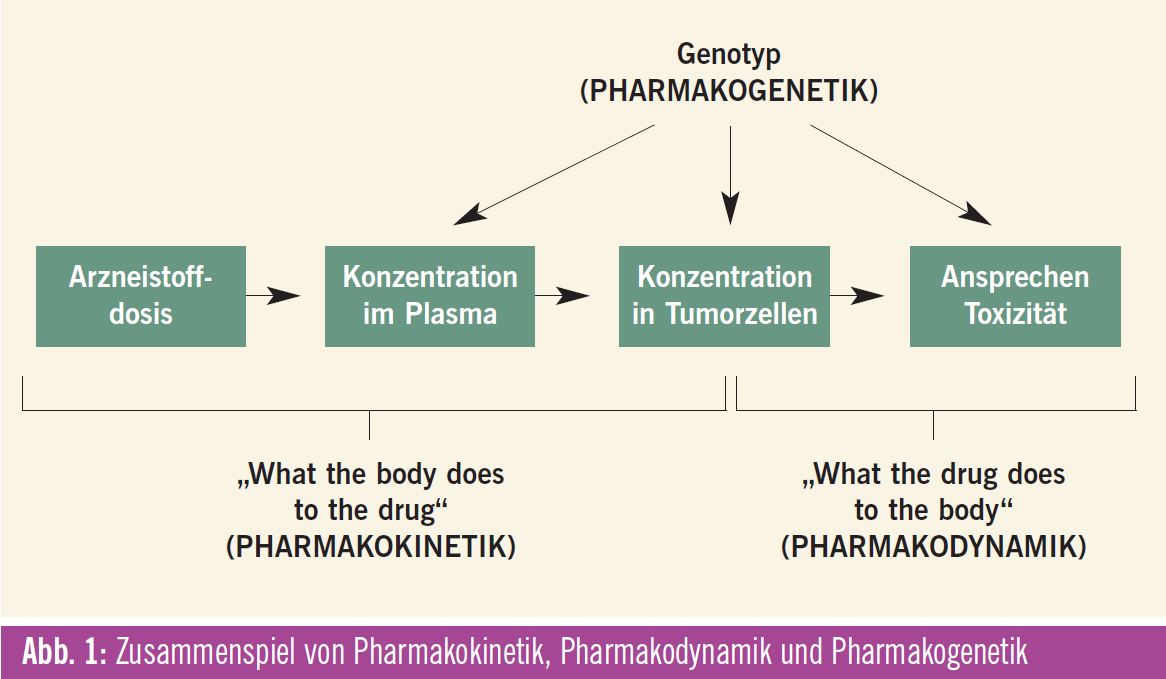 Eine rationale Dosisanpassung kann auf der Grundlage von Zusammenhängen zwischen Pharmakokinetik, Pharmakogenetik und Pharmakodynamik erfolgen (Abb. 1). Die Pharmakokinetik beschreibt die nach Applikation eines Arzneistoffes ablaufenden Prozesse im Körper (Absorption, Distribution, Metabolismus und Elimination, „ADME-Schema“). Die Pharmakodynamik wiederum beschreibt die Konzentrations-Wirkung- Beziehung am Wirkort. Mit Hilfe der Genotypisierung von Enzymen, die z. B. am Metabolismus einzelner Zytostatika beteiligt sind, können Patienten mit einem erhöhten Toxizitätsrisiko identifiziert werden.
Eine rationale Dosisanpassung kann auf der Grundlage von Zusammenhängen zwischen Pharmakokinetik, Pharmakogenetik und Pharmakodynamik erfolgen (Abb. 1). Die Pharmakokinetik beschreibt die nach Applikation eines Arzneistoffes ablaufenden Prozesse im Körper (Absorption, Distribution, Metabolismus und Elimination, „ADME-Schema“). Die Pharmakodynamik wiederum beschreibt die Konzentrations-Wirkung- Beziehung am Wirkort. Mit Hilfe der Genotypisierung von Enzymen, die z. B. am Metabolismus einzelner Zytostatika beteiligt sind, können Patienten mit einem erhöhten Toxizitätsrisiko identifiziert werden.
Tumortherapie: komplexes Zusammenspiel vieler Faktoren
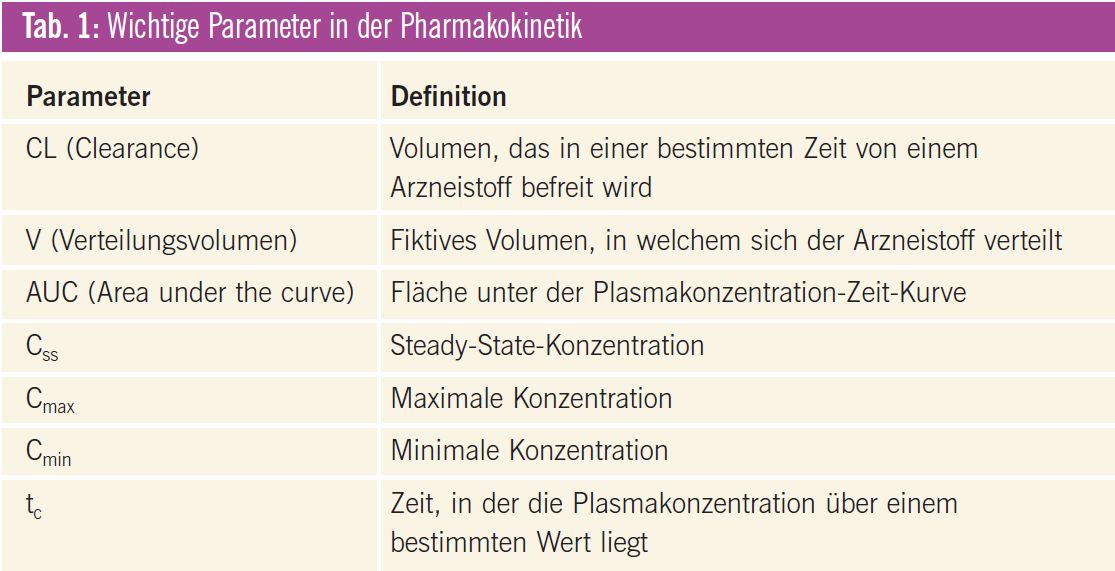 Die Tumortherapie ist meist eine Gratwanderung zwischen erwünschten (Ansprechen) und unerwünschten (Toxizität) Wirkungen. Die pharmakokinetischen Parameter wie z. B. Clearance (CL) und Verteilungsvolumen (V) der Tumortherapeutika weisen häufig eine hohe interindividuelle Variabilität auf, was dazu führt, dass die Wirkungen am einzelnen Patienten schwierig vorhersehbar und somit kaum kontrollierbar sind. Eine Übersicht über wichtige pharmakokinetische Parameter gibt Tab. 1.
Die Tumortherapie ist meist eine Gratwanderung zwischen erwünschten (Ansprechen) und unerwünschten (Toxizität) Wirkungen. Die pharmakokinetischen Parameter wie z. B. Clearance (CL) und Verteilungsvolumen (V) der Tumortherapeutika weisen häufig eine hohe interindividuelle Variabilität auf, was dazu führt, dass die Wirkungen am einzelnen Patienten schwierig vorhersehbar und somit kaum kontrollierbar sind. Eine Übersicht über wichtige pharmakokinetische Parameter gibt Tab. 1.
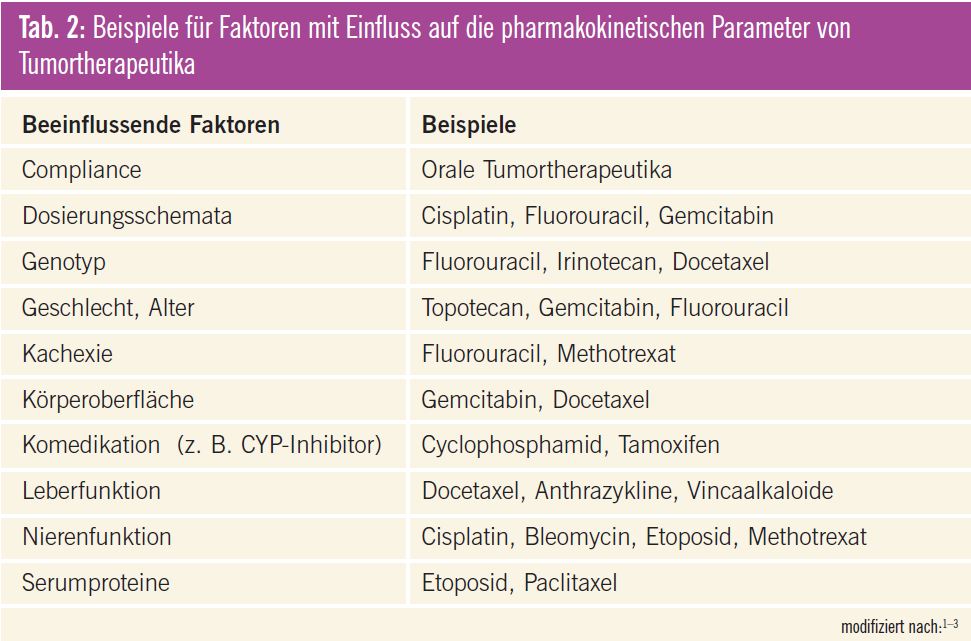
Dies hängt vor allem mit den vielfältigen Einflussfaktoren auf die Pharmakokinetik bzw. Pharmakodynamik der einzelnen Substanzen zusammen (Tab. 2). So werden immer wieder geschlechtsspezifische Unterschiede diskutiert, die allerdings von anderen Einflussfaktoren, wie dem Alter, Körpergewicht oder der Organfunktion, nur schwer zu trennen sind1–4. Um eine ausreichende Wirkung bei noch tolerierbarer Toxizität zu erzielen, ist somit eine optimale individuelle Dosisanpassung erforderlich. Erschwert wird dies allerdings dadurch, dass die einzelnen Therapien häufig nach sehr komplexen Kombinationsschemata erfolgen. Aus klinischer Sicht erfolgt aufgrund eines reduzierten Allgemeinzustandes bzw. aufgrund von eingeschränkten Organfunktionen, insbesondere bei palliativen Ansätzen, oft eine Dosisreduktion zur besseren Verträglichkeit, um eine ausreichende Lebensqualität zu erhalten. Auftretende Toxizität, insbesondere Hämatotoxizität, führt in der Praxis in der Regel ebenfalls zu einer Verminderung der Dosis oder zu einer Verlängerung der Intervalle zwischen den einzelnen Zyklen. Das Herabsetzen der Dosis aus den genannten Gründen kann jedoch zu einem Ausbleiben des Therapieerfolges führen5.
Dosisanpassung an die Körperoberfläche
 In der klinischen Routine wird bis heute die Dosis häufig an die Körperoberfläche angepasst. Hierzu wird die Formel nach Du Bois und Du Bois verwendet6: KOF (m2) = Gewicht (kg)0,425 · Größe (cm)0,725 · 0,007184 Wenn auch viele physiologische Funktionen mit der Körperoberfläche korrelieren, hängt die Clearance der meisten Tumortherapeutika nicht von der Körperoberfläche ab1–3. Daher führt diese Dosierungsstrategie bei den meisten Substanzen kaum zu einer Abnahme der Variabilität der Plasmakonzentrationen, sodass Über- bzw. Unterdosierungen hiermit nicht verhindert werden können. Ausnahmen sind z. B. Docetaxel und Gemcitabin3.
In der klinischen Routine wird bis heute die Dosis häufig an die Körperoberfläche angepasst. Hierzu wird die Formel nach Du Bois und Du Bois verwendet6: KOF (m2) = Gewicht (kg)0,425 · Größe (cm)0,725 · 0,007184 Wenn auch viele physiologische Funktionen mit der Körperoberfläche korrelieren, hängt die Clearance der meisten Tumortherapeutika nicht von der Körperoberfläche ab1–3. Daher führt diese Dosierungsstrategie bei den meisten Substanzen kaum zu einer Abnahme der Variabilität der Plasmakonzentrationen, sodass Über- bzw. Unterdosierungen hiermit nicht verhindert werden können. Ausnahmen sind z. B. Docetaxel und Gemcitabin3.
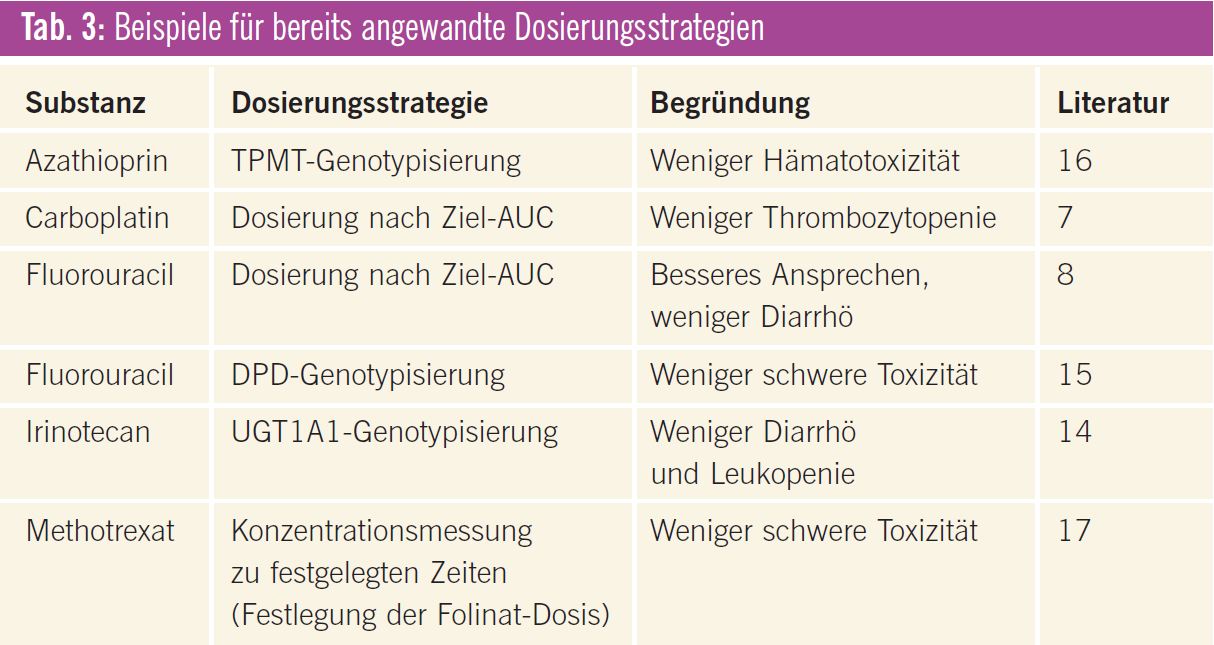
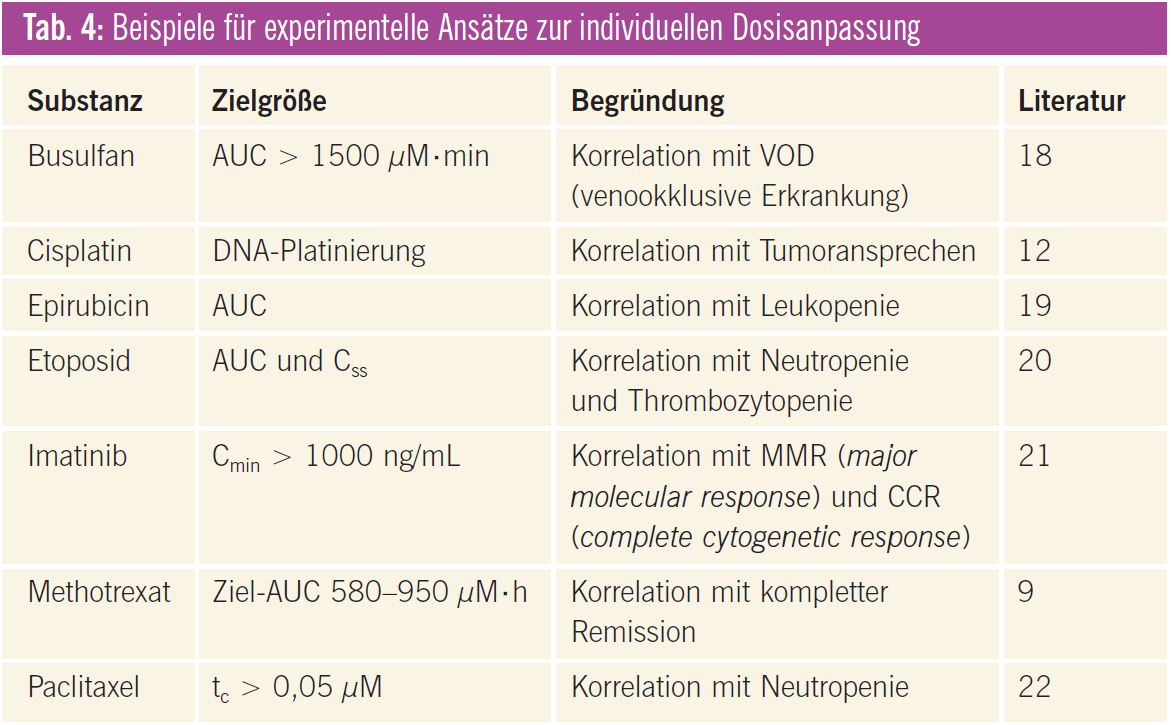
Inzwischen gibt es für einige Tumortherapeutika rationalere Dosierungsstrategien, von denen einige bereits in der klinischen Routine angewendet werden bzw. angewendet werden könnten (Tab. 3). Darüber hinaus wurden interessante Ansätze publiziert, die allerdings noch weiter untersucht werden müssen, bevor sie breit eingesetzt werden können (Tab. 4). Die wichtigsten Beispiele werden im Folgenden näher erläutert.
Pharmakokinetische Dosisanpassung
Eine Form der pharmakokinetischen Dosisanpassung stellt die Dosierung nach Ziel-AUC („Area under the curve“) dar, die heute in der Routine für Carboplatin angewendet wird. Die dosislimitierende Wirkung von Carboplatin ist das Auftreten einer Thrombozytopenie, deren Ausmaß von der Platin-AUC im ultrafiltrierbaren Plasma abhängig ist. Calvert et al. haben auf der Grundlage dieser Ergebnisse eine einfache Gleichung entwi – ckelt, mit der heute die Dosis anhand der klinisch festzulegenden Ziel-AUC und der glomerulären Filtrationsrate (GFR, meist geschätzt als Kreatinin-Clearance) leicht berechnet werden kann7:
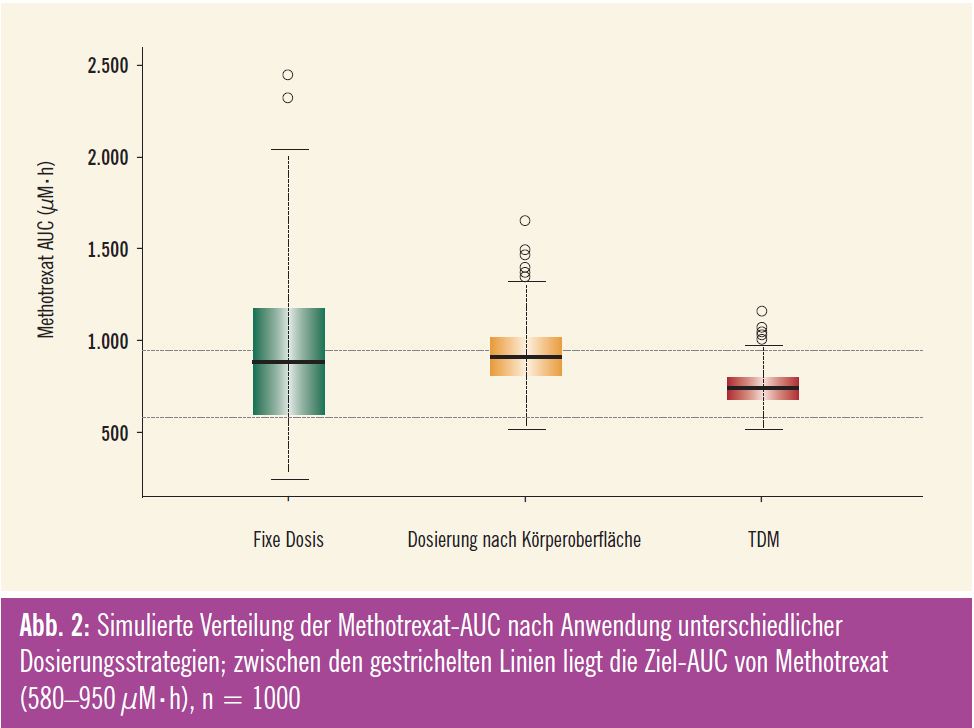
In den letzten Jahren verdichteten sich die Hinweise, dass durch eine Dosisanpassung anhand von gemessenen Plasmakonzentrationen eine höhere antitumorale Wirksamkeit und/oder eine Abnahme der Toxizität erreicht werden kann. Diese Vorgehensweise wird auch als Therapeutisches Drug Monitoring (TDM) bezeichnet. Gamelin et al. zeigten in einer prospektiven randomisierten Studie eine Verbesserung des Tumoransprechens und weniger Toxizität an Patienten mit metastasierten kolorektalen Karzinomen, bei denen die Dosis von Fluorouracil aufgrund gemessener Plasmakonzentrationen konsequent angepasst wurde, um eine vorgegebene Ziel- AUC zu erreichen8. Die Autoren berichten auch über einen Trend zu einem verlängerten Gesamtüberleben (22 Monate mit TDM gegenüber 16 Monate mit Dosierung nach Körperoberfläche). Ein anderes Beispiel ist die individuelle Dosisanpassung von Methotrexat, mit der in einer großen US-amerikanischen Studie mehr Patienten mit B-ALL in einer kompletten Remission gehalten werden konnten als mit der konventionellen Dosierung9. In Abb. 2 wird der Einfluss eines Therapeutischen Drug Monitoring auf die individuelle Arzneistoffexposition (AUC) von Methotrexat anhand von Simulationen verdeutlicht. Die Boxplots zeigen die Verteilungen der AUC-Werte von Methotrexat nach unterschiedlichen Dosierungsstrategien (fixe Dosis, Dosierung nach Körperoberfläche und TDM). Eine individuelle Dosisanpassung mittels TDM führt zu einer Reduktion der inter – individuellen Variabilität, sodass bei einem wesentlich größeren Anteil der Patienten die Ziel-AUC von 580–950 μM·h erreicht werden kann.
Pharmakodynamische Dosisanpassung
Am erfolgversprechendsten wäre es, wenn man, wie z. B. bei Antihypertensiva oder Antidiabetika, den gewünschten Effekt direkt einstellen und die Dosis aufgrund von Effektmessungen immer wieder anpassen könnte. Für Tumortherapeutika gibt es allerdings nur wenige pharmakodynamische Marker, die in der Routine wiederholt messbar sind. Daher sucht man nach Biomarkern, die mit dem Tumoransprechen korrelieren und dieses vorhersagen können. Die Auswahl möglicher Biomarker richtet sich vor allem nach dem Wirkmechanismus der Substanz.
So gibt es Hinweise, dass das Auftreten einer Neutropenie mit einem verbesserten klinischen Ansprechen einiger Zytostatika korreliert. Gurney beschreibt, dass in seiner Abteilung eine Dosissteigerung von 15–20 % erfolgt, wenn bei myelotoxischen Substanzen der Nadir der neutrophilen Granulozyten nicht unter 1,5 x 109/L liegt10, 11. Ein weiteres Beispiel für einen pharmakodynamischen Marker ist das Ausmaß der Bildung von Platin-DNA-Addukten, auf deren Bildung der zytotoxische Effekt der Platinkomplexe beruht12. Bei antiangiogenen Substanzen, z. B. Sunitinib, könnten sich der arterielle Blutdruck sowie im Plasma zirkulierende VEGF-Rezeptoren zur Dosisanpassung eignen13. Ob mit diesen Strategien tatsächlich bessere Therapieergebnisse erzielt werden können, ist jedoch noch offen.
Pharmakogenetische Dosisanpassung
Eine zunehmend wichtige Rolle in der Dosisindividualisierung spielt die Pharmakogenetik. Mit Hilfe der Genotypisierung von Enzymen, die am Metabolismus einzelner Zytostatika beteiligt sind, können Patienten mit einem erhöhten Toxizitätsrisiko identifiziert werden. Ein Beispiel ist die UDP-Glukuronosyltransferase (UGT1A1), die an der Metabolisierung von Irinotecan beteiligt ist. Patienten mit einen UGT1A1*28-Allel haben ein höheres Risiko einer Irinotecan-induzierten Neutropenie14. Die FDA hat diesen Test bereits 2005 zugelassen, in Europa findet er bisher nur wenig Anwendung. Als weitere Beispiele sind die Dihydropyrimidindehydrogenase (DPD) für die Metabolisierung von Fluorouracil und die Thiopurinmethyltransferase (TPMT) für die Metabolisierung von Azathioprin zu nennen. Auch bei diesen beiden Enzymen führt das Vorliegen bestimmter Genotypen zu einer reduzierten Enzymaktivität und hierdurch zu einer erhöhten Toxizität15, 16.
Fazit
Neben der üblichen, aber wenig hilfreichen Dosisanpassung an die Körperoberfläche der Patienten gibt es für Tumortherapeutika eine Reihe von pharmakokinetischen, pharmakodynamischen und pharmakogenetischen Dosierungsstrategien, von denen jedoch bisher nur wenige im klinischen Alltag eingesetzt werden. Häufig werden die Kosten und der Aufwand für die Messungen als Begründung angeführt, warum sich diese Strategien in der Routine nur schwer durchsetzen. Auch wenn es nur wenige prospektive Studien gibt, ist doch davon auszugehen, dass das Kosten-Nutzen- Verhältnis individueller Dosierungsstra – tegien vorteilhafter ist als das vieler neuer „targeted drugs“, für die das Gesundheits wesen viel Geld aufbringt.
FACT-BOX
Die heute für die meisten Tumortherapeutika praktizierte Dosierung nach der Körperoberfläche hat keine wissenschaftliche Grundlage. Das Potenzial pharmakokinetischer, pharmakodynamischer und pharmakogenetischer Ansätze zur individuellen Dosisanpassung wird deutlich unterschätzt. Verfügbare rationale Dosierungsstrategien sollten in Zukunft verstärkt genutzt werden, um die Tumortherapie für den einzelnen Patienten sicherer und effektiver zu gestalten.
1 Felici A et al., Eur J Cancer 2002; 38:1677–84
2 Kaestner S et al., Clin Oncol (R Coll Radiol) 2007; 19:23–37
3 Gurney H, J Clin Oncol 1996; 14:2590–611
4 Mader RM et al., Memo 2011; 4:1–5
5 Budman D, Semin Oncol 2004; 31(6 Suppl 15):3–9
6 Du Bois D et al., Arch Intern Med 1916; 17:863-71
7 Calvert A et al., J Clin Oncol 1989; 7:1748–56
8 Gamelin E et al., J Clin Oncol 2008; 26:2099–105
9 Evans W et al., N Engl J Med 1998; 338:499–505
10 Gurney H, Lancet Oncol 2005; 6:637–638
11 Di Maio M et al., Lancet Oncol 2005; 6:669–77
12 Schellens J et al., Br J Cancer 1996; 73:1569–75
13 Lindauer A et al., Clin Pharmacol Ther. 2010; 87:601–8
14 Ando Y et al., Cancer Res 2000; 60:6921–6.
15 Steiner M., J Lab Med 2006; 30:438–442
16 Black A et al., Ann Intern Med 1998; 129:716–8
17 Petros W et al., Anticancer Agents. In Burton M, Shaw et al.: Applied pharmacokinetics and pharmacodynamics, 4. Aufl., Lippincott Williams and Wilkins 2006; S. 617–636
18 Grochow L., Semin Oncol 1993; 20(4 Suppl 4):18–25
19 Danesi R et al., Clin Pharmacokinet 2002; 4:431–44
20 Bennett C et al., Cancer Res 1987; 47:1952-6
21 Picard S et al., Blood 2007; 109:3496–9
22 Joerger M et al., Clin Cancer Res 2007; 13:6410–8

Ursprünglich erschienen:
SO 05|2011
SO 05|2011