


























































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Zwischen Präklinik und Klinik – Neue therapeutische Strategien beim Melanom
23. Dezember 2011
Translationelle Onkologie und B-raf-Inhibition
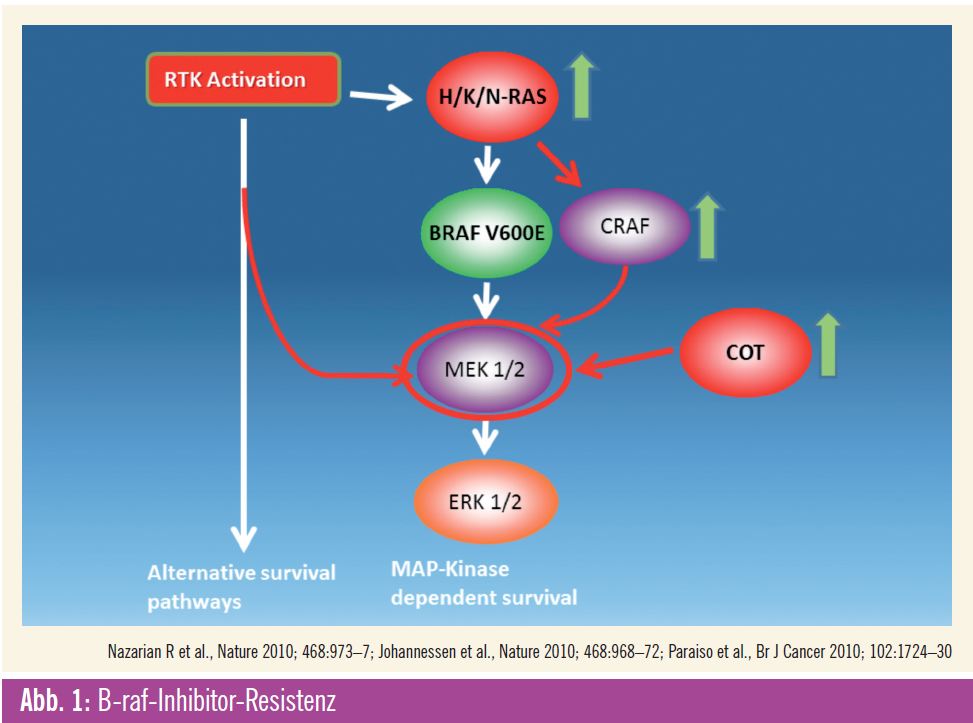 Ungefähr 50 Prozent aller Melanome weisen eine aktivierende Mutation in der im MAP-Kinase-Signalweg involvierten Kinase B-raf auf, die das Wachstum der Tumorzellen antreibt. 80 % dieser Mutationen finden sich an einer einzigen Position im Protein, eine im Vergleich zu anderen mutierten Proteinen wie z. B. c-Kit einmalige Situation. Vemurafenib ist der erste Kinaseinhibitor, der eine relative Selektivität für das mutierte Protein aufweist, und er hat in klinischen Studien eine eindrucksvolle Aktivität gezeigt. Die Braf- Kinase ist aber nur ein Teil des Signalwegs, der zur Aktivierung der mitogenaktivierten Proteinkinase (Abb. 1) führt. Bereits in frühen präklinischen Daten konnte gezeigt werden, dass zur kompletten Hemmung des Signalweges eine alleinige Hemmung der B-raf-Kinase oft nicht oder nur vorübergehend ausreichend ist1. Zusätzlich zeigten Zellkulturuntersuchungen an B-raf-mutierten Zell- Linien, die eine Resistenz gegen B-raf-Inhibitoren entwickelt hatten, dass der MAP-Kinase-Signalweg durch Aktivierung von MEK, einer B-raf nachgeschalteten Kinase, unter Umgehung einer direkten B-raf-Aktivierung aktiviert werden kann. Beschrieben wurden z. B. die Aktivierung von COT (einer alternativen Kinase), die verstärkte Expression der raf-Isoform Craf, das Auftreten von N-ras-Mutationen oder die Aktivierung von Tyrosinkinaserezeptoren, die nun als Treiber des Tumorwachstums dienen können2, 3 (Abb. 1).
Ungefähr 50 Prozent aller Melanome weisen eine aktivierende Mutation in der im MAP-Kinase-Signalweg involvierten Kinase B-raf auf, die das Wachstum der Tumorzellen antreibt. 80 % dieser Mutationen finden sich an einer einzigen Position im Protein, eine im Vergleich zu anderen mutierten Proteinen wie z. B. c-Kit einmalige Situation. Vemurafenib ist der erste Kinaseinhibitor, der eine relative Selektivität für das mutierte Protein aufweist, und er hat in klinischen Studien eine eindrucksvolle Aktivität gezeigt. Die Braf- Kinase ist aber nur ein Teil des Signalwegs, der zur Aktivierung der mitogenaktivierten Proteinkinase (Abb. 1) führt. Bereits in frühen präklinischen Daten konnte gezeigt werden, dass zur kompletten Hemmung des Signalweges eine alleinige Hemmung der B-raf-Kinase oft nicht oder nur vorübergehend ausreichend ist1. Zusätzlich zeigten Zellkulturuntersuchungen an B-raf-mutierten Zell- Linien, die eine Resistenz gegen B-raf-Inhibitoren entwickelt hatten, dass der MAP-Kinase-Signalweg durch Aktivierung von MEK, einer B-raf nachgeschalteten Kinase, unter Umgehung einer direkten B-raf-Aktivierung aktiviert werden kann. Beschrieben wurden z. B. die Aktivierung von COT (einer alternativen Kinase), die verstärkte Expression der raf-Isoform Craf, das Auftreten von N-ras-Mutationen oder die Aktivierung von Tyrosinkinaserezeptoren, die nun als Treiber des Tumorwachstums dienen können2, 3 (Abb. 1).
Basierend auf diesen Daten wurde bereits früh die Kombination eines B-raf-Inhibitors mit einem MEK-Inhibitor als mögliche Kombinationstherapie mit besserem Ansprechen und geringerem Auftreten von Resistenzen vorgeschlagen4. Erste klinische Daten aus einer Phase-I-Dosissteigerungsstudie zu dieser Kombinationstherapie wurden sowohl am letzten ASCO- Meeting in Chicago als auch beim Annual Meeting der Society of Melanoma Research in Tampa, USA vorgestellt und zeigten teils sehr hohe Ansprechraten (J Clin Oncol 29:2011 [suppl; abstr CRA8503]). In einer Kohorte von Patienten, die bereits unter B-raf-Inhibitor- Therapie eine entsprechende Resistenz entwickelt hatten, konnte durch die Kombinationstherapie wiederum ein Ansprechen auf die Therapie erzielt werden (Flaherty K et al., SMR 2011).
Ein weiterer Aspekt der Kombination aus einem B-raf- und einem MEK-Inhibitor betrifft das Auftreten von spezifischen Nebenwirkungen. Unter B-raf-Inhibitor- Therapie wird ein gehäuftes Auftreten kutaner Plattenepithelkarzinome, zumeist vom Typ des Keratoakanthoms, beobachtet. Die Arbeitsgruppe von Richard Marais konnte bereits kurz nach Bekanntwerden dieser Nebenwirkung in einem präklinischem Modell zeigen, dass eine paradoxe Aktivierung des MAPKinase- Signalwegs durch B-raf-Inhibitoren in Zellen mit einer Ras-Mutation die wahrscheinlichste Ursache ist5. Diese präklinischen Daten konnten auch in eindrucksvoller Weise „translationell“ bestätigt werden. Einerseits fanden sich in einer Serie von unter der Therapie mit Braf- Inhibitoren aufgetreten Tumoren exklusive Mutationen in H-ras, andererseits konnte durch die Kombination mit einem MEK-Inhibitor in den oben erwähnten Studien die Frequenz von Plattenepi – thelkarzinomen auf unter 1 % gesenkt werden.
Eine Auswahl therapeutischer Ansätze, welche sich in klinischer Entwicklung in der Behandlung des metastasierten Mela – noms befinden, findet sich in der Tabelle. Besonders erfolgversprechend erscheinen Kombinationen von verschiedenen Kinaseinhibitoren oder die Kombination einzelner Kinaseinhibitoren mit Immuntherapeutika.
EGF-Rezeptor-Inhibitoren in der Melanomtherapie
Die Familie der EGF-Rezeptoren (Epidermal- Growth Factor), auch HER- (Human- Epidermal Growth Factor-Rezeptors) oder ErbB-Rezeptoren (Erythroblastic Leukemia Viral Oncogene) genannt, umfasst 4 Tyrosinkinaserezeptoren. Nur zwei Mitglieder dieser Familie sind komplette Rezeptorproteine, nämlich EGFR-1 und EGFR-4. EGFR-2, besser bekannt als HER2, fehlt die Ligandenbindungsdomäne, wogegen EGFR-3 keine intrazelluläre Tyrosinkinasedomäne aufweist.
Nach Bindung des Liganden kommt es zur Bildung von Homo- und Heterodimeren der Rezeptorproteine und zur Aktivierung der Tyrosin-Kinase-Einheit. Eine Reihe von Inhibitoren dieser Rezeptoren, sowohl Antikörper als auch Kinaseinhibitoren, ist in der Therapie von Tumorerkrankungen in Verwendung6.
Auf Melanomzellen finden sich sämtliche EGF-Rezeptoren. Eine starke Expression von EGFR-1 wurde mit einer höheren Tumordicke, einer höheren Wahrscheinlichkeit für das Vorliegen eines ulzerierten Primärtumors sowie mit einer höheren Wahrscheinlichkeit für das Auftreten von Metastasen in Zusammenhang gebracht7, 8 In ähnlicher Weise konnte auch die Expression von EGFR-3 mit dem Erkrankungsstadium und dem Gesamtüberleben von Melanompatienten in Zusammenhang gebracht werden9. Aktivierende Mutationen in EGFR-1, wie sie aus anderen Tumorentitäten bekannt sind, wurden im Melanom nicht entdeckt, jedoch wurden Lapatinib-sensitive Mutationen für EGFR-4 in Melanomzellen beschrieben10. Ebenso zeigte sich in präklinischen Modellen eine Sensitivität von Melanomzellen gegenüber dem EGFR-1-Inhibitor Erlotinib. Dieser kann dabei sowohl das Tumorwachstum selbst als auch die Tumorangiogenese beeinflussen. Interessanterweise lässt sich dieser Effekt durch die Kombination mit einem VEGF-blockierenden Antikörper deutlich verstärken, wobei nicht nur der antiangiogenetische Effekt verstärkt wird, sondern in der Kombination jetzt auch die Apoptose von Tumorzellen ausgelöst wird. Darüber hinaus beeinflusst diese Therapiekombination auch das Metastasierungsverhalten von Melanomzellen in In-vivo-Modellen11. Die Bedeutung dieser Daten wurde in einem präklinischen Tumormodell eines Plattenepithelkarzinoms eindrucksvoll bestätigt und in sehr ausführlichen Untersuchungen mechanistisch belegt12; eine klinische Umsetzung ist derzeit noch ausständig.
Immuntherapien: Das Beispiel Anti-CTLA4-Antikörper
Zahlreiche Versuche einer Immuntherapie wurden gestartet und im Melanom getestet. Ein viel versprechender Ansatz ist die Verwendung von Antikörpern gegen cytotoxic T-lymphocyte-associated protein 4 (CTLA4). CTLA4 mediiert mehrere Funktionen in zytotoxischen und regulatorischen T-Zellen, welche die Immunantwort niederregulieren. Die Behandlung mit Anti-CTLA4-Antikörpern, wie zum Beispiel mit Ipilimumab (MDX- 010, Medarex Inc./Bristol-Myers Squibb, Princeton, NJ, USA) scheint die immunmediierte Zerstörung von Tumorzellen zu verstärken. Im Stadium-IV-Melanom war Ipilimumab in einer klinischen Studie erfolgreich und ist bereits in Europa und den USA zugelassen (Yervoy®). Zurzeit wird Ipilimumab im adjuvanten Setting getestet (in der EORTC-Studie 18071 [NCT00636168]), eine Phase-III-Multicenter- Studie, die in Europa und in den USA läuft). Hier wird der Effekt von adjuvantem Ipilimumab mit den Endpunkten Tumorrezidiv und Überleben mit Placebo verglichen. Inklusionskriterien sind Stadium- III-Melanome mit zumindest einer Lymphknotenmetastase größer als 1 mm. Zu erwähnen ist, dass der Anti-CTLA4- Antikörper signifikante autoimmune Toxizität verursachen kann, wie zum Beispiel Colitis, Hepatitis und Hypophysitis, daher wird es wichtig sein zu zeigen, dass in der adjuvanten Therapie die Wirkung diese Nebenwirkungen ausgleichen kann.
Präklinische Onkologie: Das Beispiel Fumarsäureester
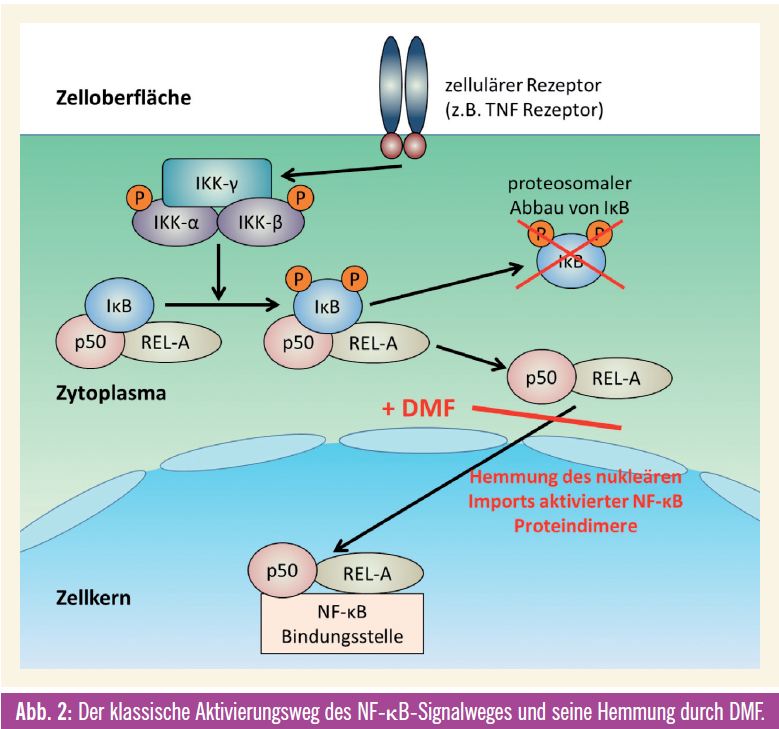
Fumarsäureester, vor allem Dimethylfumarat, werden seit Jahrzehnten erfolgreich in der medikamentösen Therapie der Psoriasis (Schuppenflechte) eingesetzt. Bis Anfang des letzten Jahrzehnts war über den Wirkmechanismus jedoch relativ wenig bekannt. Studien an kultivierten Gefäßzellen während einer simulierten Entzündungsreaktion zeigten, dass Dimethylfumarat (DMF) bestimmte Entzündungsschritte hemmen konnte. In weiteren Untersuchungen konnte eine Interaktion von DMF mit dem NF-_B-Signalweg als wesentlicher Mechanismus für die entzündungshemmenden Effekte identifiziert werden13. DMF führte zu einer Hemmung der Akkumulation von aktivierten Proteindimeren im Zellkern und damit zu einer gehemmten Signaltransduktion13 (Abb. 2). Da der NF-_B-Signalweg auch in zahlreichen humanen Tumorerkrankungen überstimuliert und aktiviert ist, lag hier die Überlegung nahe, einen therapeutischen Effekt von DMF auch in Tumorerkrankungen mit aktiviertem NF-_B-Signalweg zu untersuchen. Da auch in humanen Melanomen dieser Signalweg überaktiviert ist, wurden diese Studien vor allem mit menschlichen Melanomzellen durchgeführt. In Zellkulturexperimenten konnte durch die Gabe von DMF das Wachstum von Melanomen signifikant gehemmt werden. Therapeutisch interessant war aber noch viel mehr die Reduktion der Lymphknotenmetastasierung in verschiedenen tierexperimentellen Modellen14. Eine Kombination mit einem für die Therapie des metastasierenden Melanoms zugelassenen Chemotherapeutikum, Dacarbazin, führte zu einem synergistischem Effekt in Hinblick auf die Reduktion der Lymphknotenmetastasierung15. Im Unterschied zu den bereits in die Klinik Einzug gehaltenen Therapeutika Vemurafenib und Ipilimumab befindet sich Dimethylfumarat noch am Beginn seiner klinischen Erprobung. Aufgrund der viel versprechenden präklinischen Daten, der Kombinationsmöglichkeit und vor allem der Tatsache, dass diese Substanz bereits seit Jahrzehnten therapeutisch zugelassen ist, sollte auch ihre Wirksamkeit in der Behandlung des metastierenden Melanoms klinisch erprobt werden.
ZUSAMMENFASSEND lässt sich festhalten, dass die neuen, translationell entwickelten Therapeutika nicht nur die Behandlung des metastasierenden Melanoms revolutioniert, sondern in den letzten Jahren auch die angewandte Grundlagenforschung beflügelt haben. Dies führte zu einem besseren Verständnis zugrunde liegender molekularer und genetischer Veränderungen, was wiederum die Basis zur Entwicklung neuer Therapiestrategien bietet.
1 Paraiso KH et al., Br J Cancer 2010; 102(12):1724–30
2 Nazarian R et al., Nature 2010; 468(7326):973–7
3 Johannessen CM et al., Nature 2010; 468(7326):968–72
4 Emery CM et al., Proc Natl Acad Sci U S A 2009; 106(48):20411–6
5 Heidorn SJ et al., Cell 2010; 140(2):209–21
6 Ciardiello F, Tortora G, N Engl J Med 2008; 358(11):1160–74
7 Udart M et al., Neoplasia 2001; 3(3):245–54
8 Rakosy Z et al., Int J Cancer 2007; 121(8):1729–37
9 Reschke M et al., Clin Cancer Res 2008; 14(16):5188–97
10 Prickett TD et al., Nat Genet 2009; 41(10):1127–32
11 Schicher N et al., Clin Cancer Res 2009; 15(10):3495–502
12 Lichtenberger BM et al., Cell 2010; 140(2):268–79
13 Loewe R et al., J Immunol 2002; 168(9):4781–4787
14 Loewe R et al., Cancer Research 2006; 66(24):11888–96
15 Valero T et al., J Invest Dermatol 2010; 130(4):1087–94

Ursprünglich erschienen:
SO 05|2011
SO 05|2011