


























































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Die Prävalenz aktinischer Keratosen (AK) variiert stark je nach geografischer Lage, Hauttyp und Lebensgewohnheiten der betroffenen Populationen. In Australien, dem Land mit der höchsten UV-Belastung weltweit, weisen Studien eine Lebenszeitprävalenz von bis zu 60 % bei Personen über 60 Jahre auf. In mitteleuropäischen Ländern schwanken die Angaben zwischen 15 % und 25 % in der älteren Allgemeinbevölkerung – mit deutlich höheren Raten in Berufsgruppen mit chronischer Sonnenexposition.
Risikofaktoren
Als Hauptrisikofaktor gilt die kumulative ultraviolette Strahlenbelastung der Haut. Weitere prädisponierende Faktoren umfassen helle Hauttypen (Fitzpatrick I und II), hohes Lebensalter, männliches Geschlecht, Immunsuppression – etwa nach Organtransplantation –, genetische Disposition sowie das Vorliegen bereits bestehender Hautkrebserkrankungen. Das Risiko für die Weiterentwicklung einer AK in ein invasives Plattenepithelkarzinom der Haut (iSCC) liegt bei bis zu 10 %. Allerdings ist die Datenlage zu prognostischen Faktoren für die Transformation nicht ausreichend, um konkrete Aussagen über die Progressionswahrscheinlichkeit zu treffen. Prognostisch als ungünstig erwiesen haben sich Immunsuppression, Therapieresistenz und/oder Feldkanzerisierung. Das iSCC ist nach dem Basalzellkarzinom (BCC) der zweithäufigste nichtmelanozytäre Hauttumor (NMSC) und umfasst ca. 20 % aller NMSC.
Pathogenese und Klinik
Pathogenetisch resultieren AK aus UV-induzierten Mutationen in Tumorsuppressorgenen, insbesondere in TP53, sowie aus einer gestörten DNA-Reparatur und chronischer Entzündungsreaktion der Haut. Histologisch zeigen sich Atypien der basalen Keratinozyten mit Verlust der normalen Polarisation, Pleomorphismus und erhöhter Mitoserate – Zeichen einer intraepidermalen Dysplasie, die in schweren Fällen die gesamte Epidermisdicke erfasst.
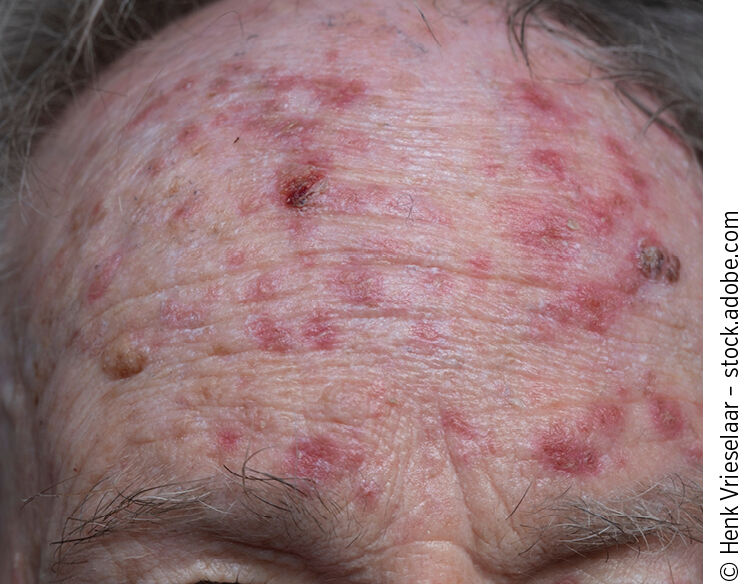
Klinisch präsentieren sich AK als raue, schuppige Erytheme oder erythematöse Plaques mit gelblich-bräunlicher, fest haftender Hyperkeratose, bevorzugt an lichtexponierten Arealen wie Gesicht, Ohren, Handrücken, Unterarmen und Kapillitium. Die Diagnosestellung erfolgt in der Regel klinisch; eine Dermatoskopie oder – bei Unsicherheiten – eine Biopsie zur histologischen Sicherung kann ergänzend eingesetzt werden.
Behandlungsmöglichkeiten
Die Therapie der AK richtet sich nach Anzahl, Lokalisation und Schweregrad der Läsionen sowie dem Allgemeinzustand der Patient:innen. Grundsätzlich wird zwischen läsionsgerichteten und feldgerichteten Therapieverfahren unterschieden. Läsionsgerichtete Verfahren eignen sich bei isolierten oder wenigen Läsionen. Die Kryotherapie mit flüssigem Stickstoff (−196 °C) gilt als Standardmethode und zeigt Abheilungsraten von 70–90 %, ist jedoch bei multiplen Herden aufwendig. Die chirurgische Exzision oder Kürettage bietet den Vorteil einer histologischen Aufarbeitung und wird bevorzugt bei hyperkeratotischen oder klinisch suspekten Läsionen eingesetzt. Feldgerichtete Therapien sind bei multiplen Läsionen oder großflächigen Kanzerisierungsfeldern indiziert. Topische Präparate wie 5-Fluorouracil (5-FU), Tirbanibulin, Diclofenac-Natrium-Gel sowie Imiquimod wirken durch unterschiedliche Mechanismen zytotoxisch oder immunmodulatorisch auf atypische Keratinozyten. Die photodynamische Therapie (PDT) mittels Aminolävulinsäure oder Methylaminolävulinat stellt eine Alternative mit besonders guten kosmetischen Ergebnissen dar.
Prävention und Nachsorge
Konsequenter Sonnenschutz durch Lichtschutzpräparate mit hohem Lichtschutzfaktor, UV-Schutzkleidung sowie die Vermeidung intensiver Sonnenexposition sind zentrale Säulen der Primärprävention. Bei Patient:innen mit multiplen oder rezidivierenden AK sowie bei immunsupprimierten Personen sind regelmäßige dermatologische Verlaufskontrollen unerlässlich, um eine Progression zu invasiven Plattenepithelkarzinomen frühzeitig zu erkennen.

Autor:
Ao. Univ.-Prof. Dr. Rainer Kunstfeld
Ao. Univ.-Prof. Dr. Rainer Kunstfeld
Universitätsklinik für Dermatologie, Medizinische Universität Wien

Ursprünglich erschienen:
AEK 12|2026
AEK 12|2026
Literatur beim Verfasser
Bildnachweis
Vorschaubild: © Kittiphan - stock.adobe.com