
























































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
In-vitro-Modelle und zelluläre Mechanismen chronischer Nierenerkrankungen
30. März 2012
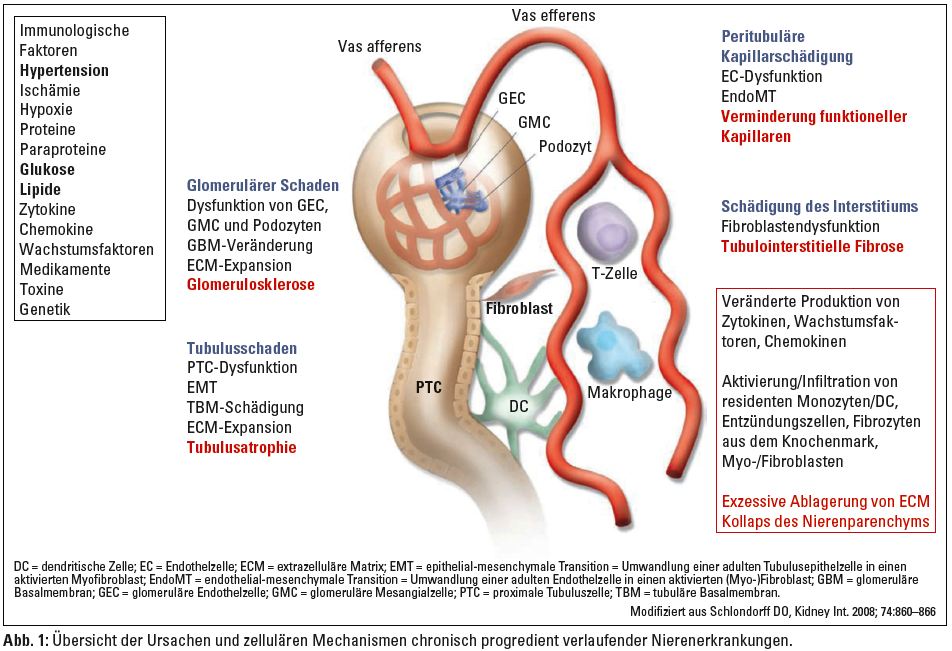 Arterielle Hypertonie, Diabetes mellitus und überhöhte Kalorienzufuhr stehen an der Spitze der Ursachen für Nierenschäden oder können diese zusätzlich zu immunologischen, infektiösen oder genetischen Faktoren verstärken. Je nach Genese und Pathomechanismus sind alle Anteile des Nierenparenchyms, also Glomeruli, Tubuli, peritubuläre Kapillaren sowie das Interstitium mehr oder weniger betroffen (Abb. 1).
Arterielle Hypertonie, Diabetes mellitus und überhöhte Kalorienzufuhr stehen an der Spitze der Ursachen für Nierenschäden oder können diese zusätzlich zu immunologischen, infektiösen oder genetischen Faktoren verstärken. Je nach Genese und Pathomechanismus sind alle Anteile des Nierenparenchyms, also Glomeruli, Tubuli, peritubuläre Kapillaren sowie das Interstitium mehr oder weniger betroffen (Abb. 1).
Tubulointerstitium wie auch Glomeruli sind am Fortschreiten chronischer Nierenerkrankungen beteiligt und prognostisch relevant.1 Chronisch progressive Nierenerkrankungen zeichnen sich durch Glomerulosklerose, tubuläre Atrophie, Infiltration von Entzündungszellen, Verminderung funktioneller Kapillaren und tubulointerstitielle Fibrose unterschiedlichen Ausmaßes aus. All diese Veränderungen sind von einer gestörten Produktion von Zytokinen, Wachstumsfaktoren und Chemokinen getragen, führen unter Mitwirkung lokaler und in das Nierenparenchym einwandernder Zelltypen zu exzessiver Produktion extrazellulärer Matrix und schließlich zum Verlust funktionellen Nierenparenchyms (Abb. 1).
Glomeruläre Zellen und Schädigungsmechanismen
Die Komplexität der Vorgänge, die in eine Nierenfibrose münden, zeigt sich unter anderem daran, dass sämtliche renale Zelltypen in der einen oder anderen Form pathogenetisch beteiligt sein können. Im Rahmen glomerulärer Insulte betrifft dies neben glomerulären Endothelzellen (GEC), Mesangialzellen (GMC) und Podozyten weiters Vorläuferzellen aus dem Knochenmark (progenitor cells) sowie infiltrierende Entzündungszellen wie Monozyten und Makrophagen (Abb. 1).1–4 Analog zu chronischen Nierenerkrankungen anderer Genese entwickeln auch Patienten mit diabetischer Nephropathie fibrotische Veränderungen sowohl der Glomeruli als auch des Tubulointerstitiums.5 Am Beginn der Progression steht dabei häufig eine glomeruläre Fibrose. Dennoch kann auch eine tubulointerstitielle Fibrose in frühen Stadien diabetischer Nierenschädigung auftreten. Meist findet sich diese jedoch zu einem späteren Zeitpunkt der Erkrankung und korreliert besser mit dem Abfall der Nierenfunktion als glomeruläre Veränderungen.5
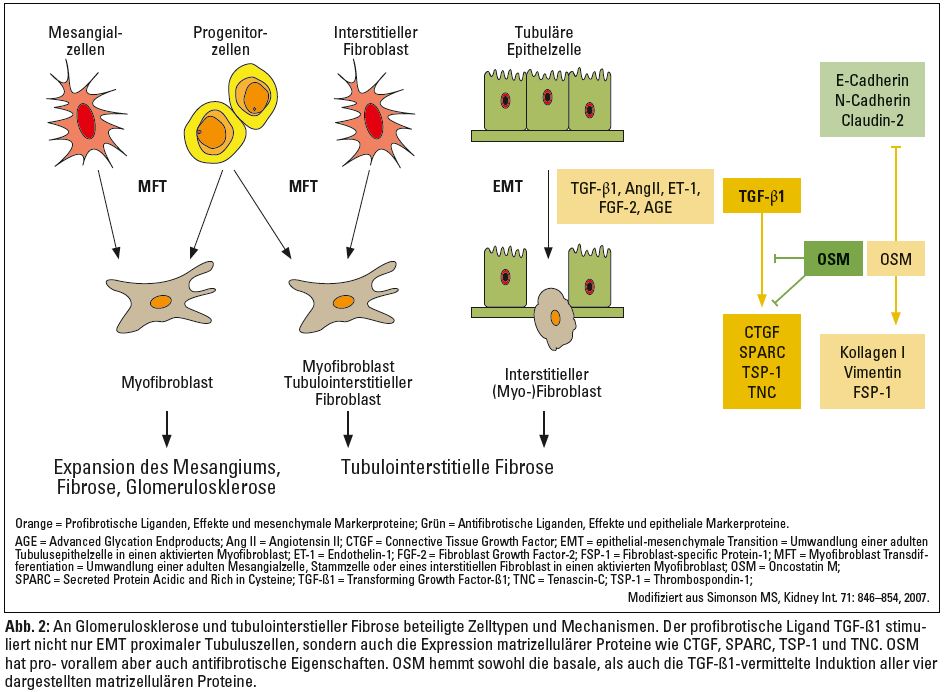 Wenngleich im Zuge von Nierenfibrosen Fibroblasten und Myofibroblasten die eigentlichen Effektorzellen darstellen, sind auch viele der anderen Nierenzellen in der Lage, ihren Phänotyp (Differenzierungsgrad) so zu verändern, dass sie zur Akkumulation und zum Umbau der extrazellulären Matrix beitragen.6 Myofibroblasten-Transdifferenzierung (MFT) und epithelial-mesenchymale Transition (EMT) stellen zwei derartige Mechanismen dar, die mit einer massiven Änderung des Differenzierungsgrades und damit der Eigenschaften der beteiligten Zellen verbunden sind (Abb. 2). Ausgelöst durch unterschiedliche Noxen und die damit einhergehende Produktion bestimmter Zytokine, Chemokine und Wachstumsfaktoren, sind sowohl glomeruläre Mesangialzellen also auch Vorläuferzellen in der Lage, sich in Myofibroblasten umzuwandeln und in der Folge vermehrt Matrixproteine (v. a. fibrilläres Kollagen und Fibronektin) zu produzieren (Abb. 2). Auf diese Weise entstandene Myofibroblasten sezernieren Proteaseinhibitoren, Zytokine, Wachstumsfaktoren sowie Entzündungsmediatoren, die diese fibrosierenden Prozesse nicht nur unterhalten, sondern weiter verstärken. MFT von Mesangialzellen (also eine Umwandlung von Mesangialzellen in aktivierte Myofibroblasten) könnte schließlich auch an der Schädigung glomerulärer Endothelzellen und von Podozyten mitwirken. Insbesondere der transforming growth factor-β (TGF-β), der von diesen dedifferenzierten Zellen (Myofibroblasten) vermehrt produziert und sezerniert wird, reguliert viele Endothelzellfunktionen wie zum Beispiel Proliferation, Migration und Apoptose. Unter dem Einfluss von TGF-ß sind auch Endothelzellen in der Lage, ihren Phänotyp in Richtung Myofibroblast zu ändern, ein Prozess, der als EndoMT (endothelial-mesenchymale Transition) bekannt ist.7 Über eine Erhöhung der Zahl glomerulärer Myofibroblasten könnte auch EndoMT an der glomerulären Fibrogenese beteiligt sein.8 Vergleichbare Änderungen der Zahl, Struktur und Funktion von Podozyten wurden bei Patienten mit Glomerulopathien und Diabetes mellitus sowie in einem transgenen Mausmodell für Glomerulosklerose nachgewiesen.9–11 Durch TGF-β, Angiotensin II (Ang II) und/oder hohe Glukosespiegel aktivierte Podozyten durchlaufen Apoptose (programmierten Zelltod) und können sich überdies von der glomerulären Basalmembran ablösen – zwei Vorgänge, die höchstwahrscheinlich an der Entwicklung einer Glomerulosklerose beteiligt sind.12 Die zur Glomerulosklerose führenden zellulären Veränderungen lassen sich schließlich folgendermaßen zusammenfassen:
Wenngleich im Zuge von Nierenfibrosen Fibroblasten und Myofibroblasten die eigentlichen Effektorzellen darstellen, sind auch viele der anderen Nierenzellen in der Lage, ihren Phänotyp (Differenzierungsgrad) so zu verändern, dass sie zur Akkumulation und zum Umbau der extrazellulären Matrix beitragen.6 Myofibroblasten-Transdifferenzierung (MFT) und epithelial-mesenchymale Transition (EMT) stellen zwei derartige Mechanismen dar, die mit einer massiven Änderung des Differenzierungsgrades und damit der Eigenschaften der beteiligten Zellen verbunden sind (Abb. 2). Ausgelöst durch unterschiedliche Noxen und die damit einhergehende Produktion bestimmter Zytokine, Chemokine und Wachstumsfaktoren, sind sowohl glomeruläre Mesangialzellen also auch Vorläuferzellen in der Lage, sich in Myofibroblasten umzuwandeln und in der Folge vermehrt Matrixproteine (v. a. fibrilläres Kollagen und Fibronektin) zu produzieren (Abb. 2). Auf diese Weise entstandene Myofibroblasten sezernieren Proteaseinhibitoren, Zytokine, Wachstumsfaktoren sowie Entzündungsmediatoren, die diese fibrosierenden Prozesse nicht nur unterhalten, sondern weiter verstärken. MFT von Mesangialzellen (also eine Umwandlung von Mesangialzellen in aktivierte Myofibroblasten) könnte schließlich auch an der Schädigung glomerulärer Endothelzellen und von Podozyten mitwirken. Insbesondere der transforming growth factor-β (TGF-β), der von diesen dedifferenzierten Zellen (Myofibroblasten) vermehrt produziert und sezerniert wird, reguliert viele Endothelzellfunktionen wie zum Beispiel Proliferation, Migration und Apoptose. Unter dem Einfluss von TGF-ß sind auch Endothelzellen in der Lage, ihren Phänotyp in Richtung Myofibroblast zu ändern, ein Prozess, der als EndoMT (endothelial-mesenchymale Transition) bekannt ist.7 Über eine Erhöhung der Zahl glomerulärer Myofibroblasten könnte auch EndoMT an der glomerulären Fibrogenese beteiligt sein.8 Vergleichbare Änderungen der Zahl, Struktur und Funktion von Podozyten wurden bei Patienten mit Glomerulopathien und Diabetes mellitus sowie in einem transgenen Mausmodell für Glomerulosklerose nachgewiesen.9–11 Durch TGF-β, Angiotensin II (Ang II) und/oder hohe Glukosespiegel aktivierte Podozyten durchlaufen Apoptose (programmierten Zelltod) und können sich überdies von der glomerulären Basalmembran ablösen – zwei Vorgänge, die höchstwahrscheinlich an der Entwicklung einer Glomerulosklerose beteiligt sind.12 Die zur Glomerulosklerose führenden zellulären Veränderungen lassen sich schließlich folgendermaßen zusammenfassen:
- Podozytopenie durch Apoptose der Podozyten und/oder deren Ablösen von der glomerulären Basalmembran;
- Vermehrung von Mesangialzellen und extrazellulärer Matrix;
- Vermehrung von (Myo-)Fibroblasten durch MFT (von Mesangialzellen), EMT (von Podozyten) und/oder EndoMT (von Endothelzellen) mit Vermehrung und Umbau der extrazellulären Matrix sowie Verdickung der Basalmembran;
- Verschluss von Kapillaren.
Tubulointerstitielle Schädigungsmechanismen und beteiligte Zelltypen
An der tubulointerstitiellen Fibrose als gemeinsamer Endstrecke praktisch aller progressiv verlaufenden chronischen Nierenerkrankungen sind ebenfalls eine Vielzahl von Mechanismen beteiligt (Abb. 1):6, 13
- Infiltration von Entzündungszellen wie Fibrozyten, Lymphozyten, Monozyten/Makrophagen, dendritische Zellen (DC) und Mastzellen;
- Tubuläre Zellapoptose und Atrophie;
- Vermehrung und Aktivierung interstitieller (Myo-)Fibroblasten unterschiedlichen Ursprungs (via EMT, EndoMT und/oder MFT);
- Produktion, Ablagerung und Umbau extrazellulärer Matrixkomponenten (ECM-Komponenten);
- Störung und Reduktion der peritubulären Mikrozirkulation.
Schädigung von Nierengewebe induziert häufig entzündliche Veränderungen und Infiltration von Entzündungszellen, wie Fibrozyten, Lymphozyten, Monozyten/Makrophagen, dendritische Zellen und Mastzellen. Obwohl Entzündungsvorgänge grundsätzlich als Schutz- beziehungsweise Abwehrmechanismus angesehen werden können, stellen diese, wenn sie nicht selbstlimitierend wirken, einen wichtigen Initialfaktor für die Entwicklung einer Fibrose dar. Entzündungsmechanismen bei anhaltender Noxe bilden daher auch die Grundlage tubulointerstitieller Fibrogenese. Fibrozyten stellen einen der beteiligten Zelltypen dar, der extrazelluläre Matrix produziert und bei einer Schädigung die Nieren entlang eines Chemokingradienten infiltriert. Alternativ können derartige fibroblastenähnliche Zellen auch lokal im entzündlichen Gewebe aus Leukozyten (de-)differenzieren.4 Der genaue Beitrag der Fibrozyten an der renalen Fibrogenese in vivo ist unsicher, da bis dato spezifische Marker dieses Zelltyps fehlen und somit eine klare Unterscheidung von Monozyten, Makrophagen und Fibroblasten oft schwer fällt. Auch zur Beteiligung von Monozyten, Makrophagen, dendritischen Zellen und Mastzellen an diesen initialen Vorgängen liegen uneinheitliche Ergebnisse vor. Hinweise auf profibrotische Funktionen von Monozyten und/oder Makrophagen stammen aus dem Modell der unilateralen uretheralen Obstruktion (UUO) in Mäusen. Ein von Makrophagen sezernierter, profibrotischer Mediator ist Galectin-3, ein β-Galactosidase-bindendes Lectin. Galectin-3-defiziente Mäuse waren bei unveränderter Makrophagenzahl gegen UUO-induzierte Fibrose geschützt.14
Ob Makrophagen tatsächlich Nierenfibrose positiv oder negativ beeinflussen, müssen weitere Untersuchungen und eine genauere Charakterisierung dieser heterogenen Zellpopulation zeigen. Ähnliches gilt auch für dendritische Zellen, die in großer Zahl im gesunden Niereninterstitium vorkommen und möglicherweise eine Rolle beim Übergang einer glomerulären Schädigung in Richtung Tubulointerstitium spielen.15 Im Gegensatz zu dendritischen Zellen sind Mastzellen in gesunden Nieren kaum zu finden, nehmen aber mit zunehmender Fibrosierung und abnehmender Nierenfunktion in ihrer Zahl zu.16, 17 Dennoch ist auch ihre Rolle im Zuge der Entwicklung einer Nierenfibrose nicht endgültig geklärt.
Fibroblasten und Myofibroblasten als eigentliche Effektorzellen
Die für die tubulointerstitielle Fibrogenese vermutlich wichtigsten Zelltypen stellen jedoch Fibroblasten und Myofibroblasten dar. Auch ihre genaue funktionelle Einordnung und Charakterisierung im Verlauf chronisch progredienter Nierenerkrankungen gestaltet sich durch die Heterogenität der aus ihnen hervorgehenden Zellpopulationen schwierig. Faktum ist, dass neben mechanischem Stress, verschiedenen Zytokinen und Wachstumsfaktoren auch viele der in Abbildung 1 (linke Box) zusammengefassten Ursachen chronischer Nierenerkrankungen in der Lage sind, sowohl die Aktivierung und Vermehrung lokal ansässiger tubulointerstitieller Fibroblasten, als auch die Bildung von Myofibroblasten unterschiedlicher Herkunft zu induzieren. Interstitielle Myofibroblasten können grundsätzlich aus aktivierten, sich dedifferenzierenden Fibroblasten hervorgehen (MFT), sich aus tubulären Epithelzellen (EMT) oder Endothelzellen (EndoMT) entwickeln beziehungsweise aus Stammzellen gebildet werden (Abb. 2).
Ein besonderes Charakteristikum dieser Myofibroblasten liegt in ihren kontraktilen Eigenschaften, die auf der De-novo-Expression von α-smooth muscle actin (α-SMA), einem kontraktilen Protein, das unter physiologischen Bedingungen nur in glatten Muskelzellen vorkommt, basieren und für die gesteigerte Motilität dieser Zellen verantwortlich sind. Weitere Charakteristika dedifferenzierter und aktivierter (Myo-)Fibroblasten sind eine gesteigerte Expression mesenchymaler Markerproteine (Vimentin, Kollagen I, FSP-1) sowie die verminderte Expression epithelialer Markerproteine (E-cadherin, N-cadherin, Claudin-2, ZO-1; Abb. 2). Unterschiedliche Auffassungen bestehen insbesondere hinsichtlich des Anteils jener Myofibroblasten, die sich in vivo durch EMT aus adulten tubulären Epithelzellen entwickeln, die tubuläre Basalmembran durchwandern, um schließlich in das Interstitium zu migrieren und auf diese Weise einen Beitrag zur tubulointerstitiellen Fibrose leisten. Gesichert ist, dass vor allem Liganden wie TGF-β 1, Ang II, EGF und FGF-2, aber auch hohe Glukosespiegel (Advanced glycation endproducts, AGE) oder monoklonale Leichtketten, wie sie beim multiplen Myelom auftreten, in vitro eine vollständige (TGF-β1) oder teilweise Umwandlung adulter tubulärer Epithelzellen zu aktivierten Myofibroblasten bewirken (Abb. 2). Unter den genannten Liganden kann TGF-β1 als der wichtigste Mediator im Zusammenhang mit der Genese chronischer Nierenerkrankungen hervorgehoben werden. TGF-β1 ist an praktisch allen profibrotischen Mechanismen beteiligt und stimuliert in erster Linie die Ausbildung und Proliferation von Fibroblasten und Myofibroblasten sowie die exzessive Produktion und Ablagerung extrazellulärer Matrix. Auch subtilere Änderungen des tubulär-epithelialen Zellphänotyps leisten einen suffizienten Beitrag zur tubulointerstitiellen Schädigung. TGF-β1 induziert zum Beispiel die Expression sogenannter matrizellulärer Proteine wie zum Beispiel CTGF (Connective Tissue Growth Factor), SPARC (Secreted Protein Acidic Rich in Cysteine), TSP-1 (Thrombospondin-1) und TNC (Tenascin-C) in humanen proximalen Tubuluszellen (Abb. 2).18 Diese matrizellulären Proteine repräsentieren eine Gruppe extrazellulärer Matrixproteine, die selbst keine strukturellen Aufgaben besitzen, die aber andere Matrixbestandteile, membranständige Zellrezeptoren sowie Zytokine und Wachstumsfaktoren binden und auf diese Weise die Zusammensetzung der extrazellulären Matrix beeinflussen. Ihre Expression ist in gesundem adulten Gewebe gering, findet sich bei Gewebsschädigung und Wundheilungsprozesses jedoch stark hochreguliert. In Nieren scheinen zahlreiche dieser matrizellulären Proteine an der tubulointerstitiellen Fibrogenese und der Progression chronischer Nierenerkrankungen beteiligt zu sein.18
Oncostatin M: Pro- oder antifibrotisches Zytokin?
Neben den oben genannten profibrotisch wirkenden Zytokinen und Wachstumsfaktoren ist zuletzt ein weiteres Zytokin in den Mittelpunkt des Interesses gerückt: Oncostatin M (OSM), ein Mitglied der Interleukin-6-Familie, das vor allem von aktivierten T-Zellen, Monozyten und Neutrophilen sezerniert wird. Dieses Zytokin wurde ursprünglich als Proliferationshemmer verschiedener Tumorzellen beschrieben, besitzt jedoch zahlreiche weitere biologische Funktionen im Rahmen von Inflammation, Remodeling der extrazellulären Matrix, Hämatopoese, Organentwicklung und Regeneration. In Nieren wurde OSM kürzlich als Hauptmediator einer renal tubulären Akute-Phase-Antwort beschrieben und dürfte pro-, vor allem aber auch antifibrotische Funktionen besitzen.18–20 OSM ist in der Lage, die Expression epithelialer Zellkontaktproteine wie zum Beispiel von E-Cadherin, N-Cadherin und Claudin-2 in humanen proximalen Tubuluszellen zu hemmen und gleichzeitig die Expression mesenchymaler Markerproteine wie zum Beispiel von Kollagen Typ I, Vimentin und fibroblastenspezifischen Protein-1 (FSP-1) zu stimulieren (Abb. 2).20 OSM bewirkt somit in proximalen Tubuluszellen Veränderungen, die jenen einer EMT proximaler Tubuluszellen ähneln. Im Gegensatz dazu scheint OSM aber auch wichtige antifibrotische Eigenschaften zu besitzen: Während TGF-β1 einen starken Stimulus für die Expression matrizellulärer Proteine in proximalen Tubuluszellen darstellt, hemmt OSM sowohl die basale als auch die TGF-β1-vermittelte Expression der vier oben genannten matrizellulären Proteine CTGF, SPARC, TSP-1 und TNC deutlich (Abb. 2). In Abhängigkeit vom zellulären Mikromilieu und dem spezifischen Schädigungsmuster (Dauer, Schweregrad, Zellkontext, involvierte Liganden) könnte OSM also einerseits zur Schädigung und Progression, andererseits auch zum Schutz und zur Stabilisierung chronischer Nierenerkrankungen beitragen.
Grundsätzlich ist zu vermuten, dass in den von Noxen betroffenen Nierenzellen, so auch in proximalen Tubuluszellen, je nach Ausmaß, Art und Dauer einer Schädigung regelmäßig Schädigungs- und Schutzmechanismen gleichzeitig ablaufen. Welche Vorgänge unter spezifischen Umständen überwiegen, entscheidet schließlich darüber, ob eine Nierenerkrankung progredient oder stabil verläuft oder ob gar eine Reparatur möglich ist. In jedem Fall versprechen Untersuchungen, die sich mit der Plastizität der beteiligten Zelltypen und mit den zu Grunde liegenden molekularen Mechanismen beschäftigen, weitere Fortschritte bei den Bemühungen, das chronisch erkrankte Nierengewebe zu schützen oder gar zur Heilung zu bringen.
1 Schlondorff DO, Kidney Int 2008; 74:860–866
2 El Nahas AM and Bello AK, Lancet 2005; 365:331–340
3 Lee SB and Kalluri R, Kidney Int 2010; 78(Suppl. 119):S22–S26
4 Boor P et al., Nature Rev Nephrol 2010; 6:643–656
5 Simonson MS, Kidney Int 2007; 71:846–854
6 Boor P et al., Nature Rev Nephrol 2010; 6:643–656
7 Arciniegas E et al., J Cell Sci 1992; 103:521–529
8 Zeisberg EM et al., J Am Soc Nephrol 2008; 19:2282–2287
9 Srivastava T et al., Kidney Int 2001; 59:118–125
10 Steffes MW et al., Kidney Int 2001; 59:210–42113
11 Shih NY et al., Science 1999; 286:312–315
12 López-Hernández FJ and López-Novoa JM, Cell Tissue Res 2012; 347:141–154
13 Liu Y, Nature Rev Nephrol 2011; 7:684–696
14 Henderson NC et al., Am J Pathol 2008; 172:288–298
15 Heymann F et al., J Clin Invest 2009; 119:1286–1297
16 El-Koraie AF et al., Kidney Int 2001; 60:167–172
17 Kondo S et al., J Am Soc Nephrol 2001; 12:1668–1676
18 Sarközi R et al., Am J Physiol Renal Physiol 2011; 301:F1014–F1025

Ursprünglich erschienen:
Neph 01|2012
Neph 01|2012