
























































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Omics-Technologien zur Analyse von chronischen Nierenerkrankungen
30. März 2012
Seit Mitte der 1990er-Jahre kommt es zu wesentlichen Veränderungen in der biomedizinischen Forschung, die nun eine komplexe, integrative Datenanalyse und Interpretation ermöglicht. Es wurden experimentelle Analysen zur simultanen Erfassung von Veränderungen einer Vielzahl von Molekülen eines zellulären Systems entwickelt, und systembiologische Analysemethoden ermöglichen in einem gegebenen biologischen System die Erstellung eines integrierten Bildes möglichst aller regulatorischer Prozesse auf mehreren Ebenen (RNA, Proteine, Metabolite). Diese Methoden werden für gewöhnlich unter dem Begriff „Omics“ zusammengefasst, angelehnt an den ersten in diesem Zusammenhang verwendeten Begriff „Genomics“, bei dem die Genexpression (RNA) des gesamten Genoms aus nahezu jeder Art von Gewebe oder Körperflüssigkeit gleichzeitig erfasst wird. In der letzten Zeit hat der Terminus „Transcriptomics“ den alten Begriff „Genomics“ zunehmend abgelöst, um Verwechslungen mit DNA-Analysen („Genetics“ und häufig eben auch „Genomics“) zu vermeiden. Auch die gleichzeitige Messung vieler Proteine („Proteomics“) und Metaboliten („Metabolomics“) in Zusammenhang mit chronischen Nierenerkrankungen soll in diesem Artikel erläutert werden.
Transcriptomics
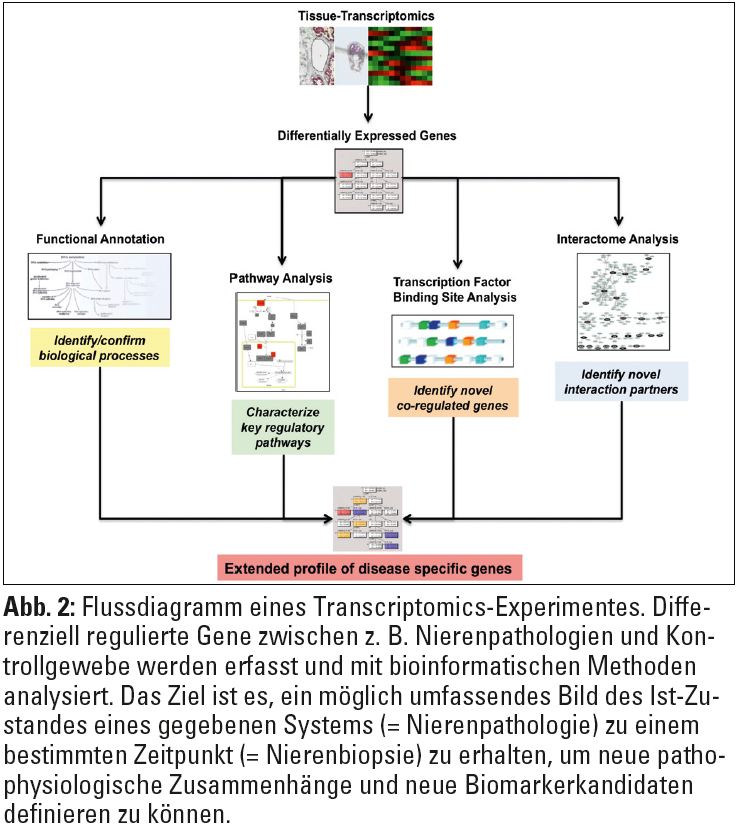
Um die Konzentration nicht nur eines einzelnen spezifischen RNA-Transkripts, sondern möglichst die RNA des gesamten Genoms in einem Hochdurchsatzverfahren („High-throughput Transcriptomics“) simultan messen zu können, werden am häufigsten so genannte Microarrays verwendet. Diese Mikroarrays bestehen aus kleinen Kassetten (ca. 3 x 6 cm) oder Glasobjektträgern (ca. 2,5 x 7,5 cm), die Oligonukleotidsequenzen nahezu aller bekannten Gene enthalten. Die neuesten Generationen der Mikroarrays ermöglichen die Analyse der Expression von 20.000 bis 30.000 Genen gleichzeitig (Abb. 1). 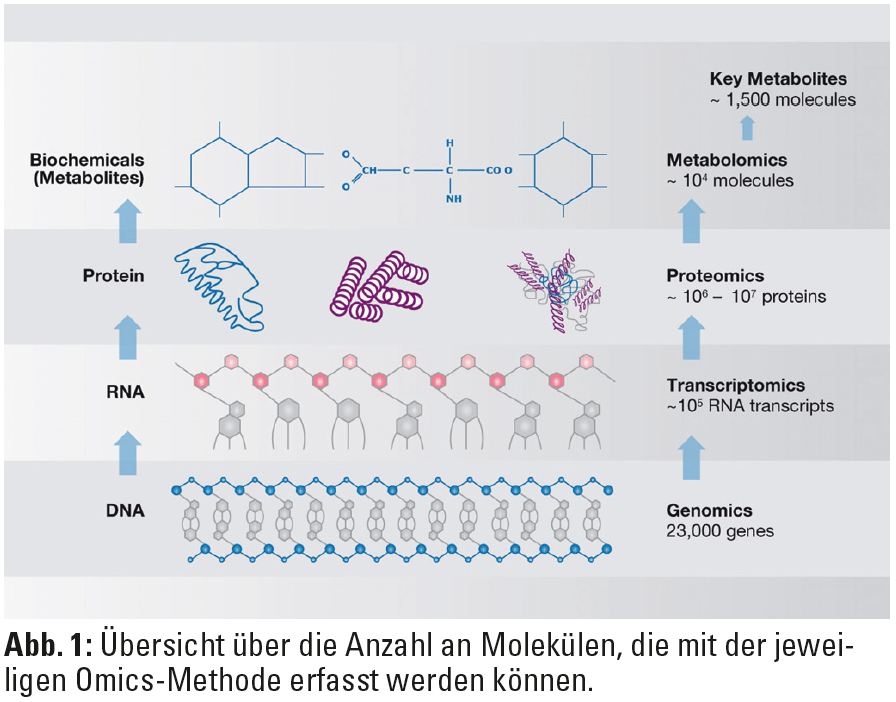 Die RNA-Moleküle sind mit Fluoreszenzfarbstoffen markiert, und die Signalintensität einzelner „spots“ wird optisch durch Laserabtastung gemessen. Diese immensen Datenmengen können nur mithilfe verschiedener, größtenteils öffentlich zugänglicher integrativer Datenbanken und Softwarelösungen systembiologisch analysiert oder mit anderen publizierten Daten in Verbindung gebracht werden. Die in diesen Datenbanken gespeicherten Informationen über die biologischen Funktionen eines bestimmten Moleküls X, bzw. über molekulare Prozesse, an denen das Molekül teilnimmt, werden dann mit geeigneten Applikationen im Sinne einer Pathway-Analyse abgerufen. So können vorher homogen erscheinende Krankheitsbilder in molekulare Untergruppen eingeteilt werden, die sich unter Umständen im klinischen Verlauf unterscheiden. Transcriptomics-Daten können auch zur Identifikation von diagnostischen und prognostischen Biomarkern eingesetzt werden (Abb. 2).
Die RNA-Moleküle sind mit Fluoreszenzfarbstoffen markiert, und die Signalintensität einzelner „spots“ wird optisch durch Laserabtastung gemessen. Diese immensen Datenmengen können nur mithilfe verschiedener, größtenteils öffentlich zugänglicher integrativer Datenbanken und Softwarelösungen systembiologisch analysiert oder mit anderen publizierten Daten in Verbindung gebracht werden. Die in diesen Datenbanken gespeicherten Informationen über die biologischen Funktionen eines bestimmten Moleküls X, bzw. über molekulare Prozesse, an denen das Molekül teilnimmt, werden dann mit geeigneten Applikationen im Sinne einer Pathway-Analyse abgerufen. So können vorher homogen erscheinende Krankheitsbilder in molekulare Untergruppen eingeteilt werden, die sich unter Umständen im klinischen Verlauf unterscheiden. Transcriptomics-Daten können auch zur Identifikation von diagnostischen und prognostischen Biomarkern eingesetzt werden (Abb. 2).
 Auf dem Gebiet der chronischen Nierenerkrankungen wurden Transcriptomics-Methoden sehr häufig für die Analyse der Genexpression in ganzen Nierenbiopsien oder in einzelnen mikrodissezierten Kompartimenten der Niere angewandt. So konnte nachgewiesen werden, dass Glomerula bei Lupus nephritis bzw. bei fokal-segmentaler Glomerulosklerose, die histologisch und auch klinisch nicht zu unterscheiden waren, doch deutliche Unterschiede in der Aktivierung verschiedener Inflammations- und Fibrosekaskaden zeigen. Bei Patienten mit diabetischer Nephropathie waren die Genexpressionsmuster des tubulointerstitiellen Kompartments bei Patienten mit einem progredienten Verlauf der Erkrankung durch eine deutliche Aktivierung von inflammationsabhängigen Genen gekennzeichnet, die mit einem spezifischen NF-kappaB-Modul assoziiert waren. Eine weiterführende Analyse zeigte dann die Aktivierung des Jak-Stat-Pathways bei progressiver, nicht aber bei stabiler diabetischer Nephropathie. Durch Transcriptomics-Analysen konnte schließlich auch die Aktivierung der Hypoxie-Respons- und VEGF-Pathways bei progressiven proteinurischen Nierenerkrankungen gezeigt werden. Transcriptomics-Methoden in Kombination mit systembiologischen Analysen haben ein vielversprechendes Potenzial zur Identifikation neuer krankheits- und gewebespezifischer Biomarker. So konnte gezeigt werden, dass nicht nur die tubulointerstitielle Inflammation einen ungünstigen Marker für die Prognose der chronischen Nierenerkrankung darstellt. Es konnten auch bestimmte Gene als Prädiktoren der Nierenfunktion identifiziert werden, deren Vorhersagekraft deutlich besser ist als herkömmliche Marker wie z. B. Kreatinin, Proteinurie oder das Ausmaß des histologischen Schadens.
Auf dem Gebiet der chronischen Nierenerkrankungen wurden Transcriptomics-Methoden sehr häufig für die Analyse der Genexpression in ganzen Nierenbiopsien oder in einzelnen mikrodissezierten Kompartimenten der Niere angewandt. So konnte nachgewiesen werden, dass Glomerula bei Lupus nephritis bzw. bei fokal-segmentaler Glomerulosklerose, die histologisch und auch klinisch nicht zu unterscheiden waren, doch deutliche Unterschiede in der Aktivierung verschiedener Inflammations- und Fibrosekaskaden zeigen. Bei Patienten mit diabetischer Nephropathie waren die Genexpressionsmuster des tubulointerstitiellen Kompartments bei Patienten mit einem progredienten Verlauf der Erkrankung durch eine deutliche Aktivierung von inflammationsabhängigen Genen gekennzeichnet, die mit einem spezifischen NF-kappaB-Modul assoziiert waren. Eine weiterführende Analyse zeigte dann die Aktivierung des Jak-Stat-Pathways bei progressiver, nicht aber bei stabiler diabetischer Nephropathie. Durch Transcriptomics-Analysen konnte schließlich auch die Aktivierung der Hypoxie-Respons- und VEGF-Pathways bei progressiven proteinurischen Nierenerkrankungen gezeigt werden. Transcriptomics-Methoden in Kombination mit systembiologischen Analysen haben ein vielversprechendes Potenzial zur Identifikation neuer krankheits- und gewebespezifischer Biomarker. So konnte gezeigt werden, dass nicht nur die tubulointerstitielle Inflammation einen ungünstigen Marker für die Prognose der chronischen Nierenerkrankung darstellt. Es konnten auch bestimmte Gene als Prädiktoren der Nierenfunktion identifiziert werden, deren Vorhersagekraft deutlich besser ist als herkömmliche Marker wie z. B. Kreatinin, Proteinurie oder das Ausmaß des histologischen Schadens.
Proteomics
Proteomics ist die quantitative und qualitative Untersuchung von Proteinen im großen Maßstab. Obwohl Proteine im Endeffekt Genprodukte darstellen, kann ein und dasselbe Gen zu verschiedenen Proteinisoformen führen. Zusätzlich kommt es durch posttranslationelle Veränderungen zu Modifikationen am Proteinmolekül, die entscheidend für die Funktion sind, jedoch durch die Messung von RNA-Konzentrationen (siehe oben) nicht erfasst werden. Eine der am längsten bekannten Proteomics-Analysemethoden ist die zweidimensionale Polyacrylamidgelelektrophorese (2D-PAGE), bei der die einzelnen Proteine einer Probe zunächst nach dem isoelektrischen Punkt und danach nach der Molekülgröße aufgetrennt werden. Heutzutage stellt die Kapillarelektrophorese (CE) eine Weiterentwicklung dieser Methode dar. Die Proteinspots können enzymatisch degradiert und mithilfe der Massenspektrometrie (MALDI-TOF, Matrix Assisted Laser Desorption Ionization – Time Of Flight) identifiziert werden (Abb. 3). 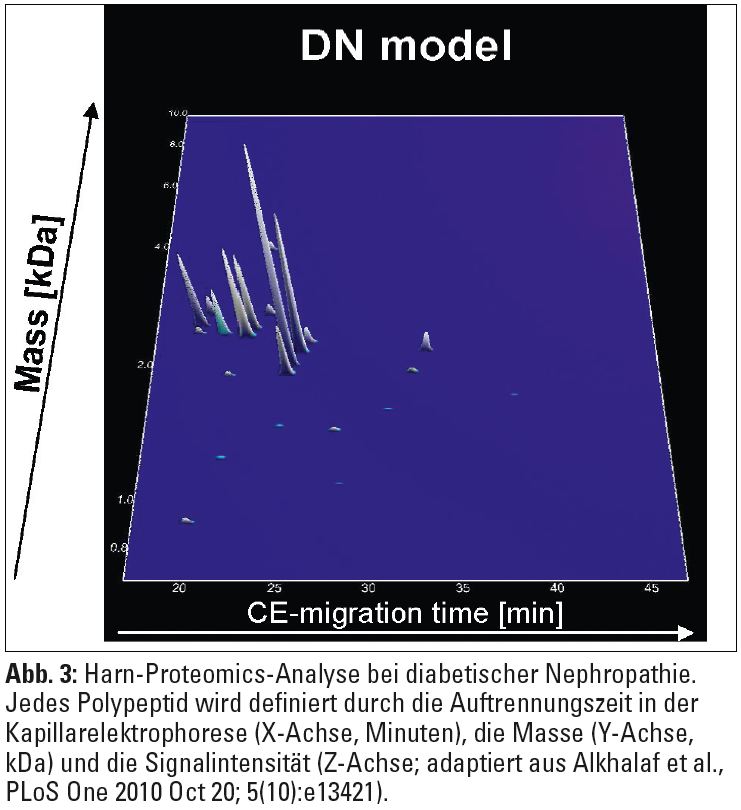 Körperflüssigkeiten wie Urin, Plasma, Liquor, Gelenksflüssigkeit oder Speichel beinhalten entweder unter physiologischen Bedingungen oder bei spezifischen Erkrankungen Tausende unterschiedliche Peptide und Proteine und eignen sich somit optimal für Proteomics-Analysen. Da die hohen Albuminkonzentrationen im Serum bzw. im Plasma eine nicht unbeträchtliche technische Hürde für die Proteomics-Analysen darstellen, hat sich die Proteomics-Forschung insbesondere bei chronischer Niereninsuffizienz vor allem auf den Urin konzentriert. So konnte gezeigt werden, dass Patienten mit unterschiedlichen chronischen Nierenerkrankungen im Vergleich zu gesunden Kontrollpersonen weniger Kollagenabbauprodukte im Harn aufweisen, sodass bei Patienten mit chronischer Niereninsuffizienz bereits in sehr frühen Stadien der Erkrankung eine Störung der Synthese und des Abbaues der extrazellulären Matrix vorliegen könnte. Im Urin von Diabetespatienten mit Mikroalbuminurie wurden Peptide nachgewiesen, die prognostische Bedeutung für die diabetische Nephropathie hatten. Interessanterweise konnte mittels Urin-Proteomics nicht nur eine Vielzahl potenzieller Biomarker für die Aktivität einer ANCA-assoziierten Nephropathie identifiziert werden, sondern es zeigte sich auch, dass eine immunsuppressive Therapie das Urin-Proteom wieder in Richtung „Remission“ verändern kann. Weitere Urin-Proteomics-Daten gibt es auch zu polyzystischen und pädiatrischen Nierenkrankheiten, urologischen Pathologien und anderen. Eine besonders vielversprechende Anwendung von Urin-Proteomics ist die Analyse von Urin-Peptiden im Harn von Patienten mit koronarer Herzkrankheit (KHK) zu diagnostischen Zwecken. Erste Daten zeigen, dass bei symptomatischen Patienten anhand von Urin-Proteom-Profilen eine vaskuläre Kalzifizierung der Koronarien mit hoher Sensitivität und Spezifität vorausgesagt werden kann.
Körperflüssigkeiten wie Urin, Plasma, Liquor, Gelenksflüssigkeit oder Speichel beinhalten entweder unter physiologischen Bedingungen oder bei spezifischen Erkrankungen Tausende unterschiedliche Peptide und Proteine und eignen sich somit optimal für Proteomics-Analysen. Da die hohen Albuminkonzentrationen im Serum bzw. im Plasma eine nicht unbeträchtliche technische Hürde für die Proteomics-Analysen darstellen, hat sich die Proteomics-Forschung insbesondere bei chronischer Niereninsuffizienz vor allem auf den Urin konzentriert. So konnte gezeigt werden, dass Patienten mit unterschiedlichen chronischen Nierenerkrankungen im Vergleich zu gesunden Kontrollpersonen weniger Kollagenabbauprodukte im Harn aufweisen, sodass bei Patienten mit chronischer Niereninsuffizienz bereits in sehr frühen Stadien der Erkrankung eine Störung der Synthese und des Abbaues der extrazellulären Matrix vorliegen könnte. Im Urin von Diabetespatienten mit Mikroalbuminurie wurden Peptide nachgewiesen, die prognostische Bedeutung für die diabetische Nephropathie hatten. Interessanterweise konnte mittels Urin-Proteomics nicht nur eine Vielzahl potenzieller Biomarker für die Aktivität einer ANCA-assoziierten Nephropathie identifiziert werden, sondern es zeigte sich auch, dass eine immunsuppressive Therapie das Urin-Proteom wieder in Richtung „Remission“ verändern kann. Weitere Urin-Proteomics-Daten gibt es auch zu polyzystischen und pädiatrischen Nierenkrankheiten, urologischen Pathologien und anderen. Eine besonders vielversprechende Anwendung von Urin-Proteomics ist die Analyse von Urin-Peptiden im Harn von Patienten mit koronarer Herzkrankheit (KHK) zu diagnostischen Zwecken. Erste Daten zeigen, dass bei symptomatischen Patienten anhand von Urin-Proteom-Profilen eine vaskuläre Kalzifizierung der Koronarien mit hoher Sensitivität und Spezifität vorausgesagt werden kann.
Metabolomics
Metabolomics ist die systematische Identifizierung und Quantifizierung aller nachweisbaren kleinen Moleküle (= Metabolite) in einem bestimmten zellulären Kompartiment, einer Zelle, in Gewebe oder in Körperflüssigkeiten (Abb. 1). Metabolite sind die funktionalen Endpunkte von physiologischen und pathophysiologischen Prozessen, und das Metabolom beschreibt die Gesamtheit aller Metabolite eines Organismus zu einem bestimmten Zeitpunkt. In den Konzentrationen der Metabolite oder deren Änderungen spiegeln sich sowohl genetische Veranlagungen als auch äußere Einflüsse wie Ernährung, Bewegung oder Medikamente wider, was unmittelbare Rückschlüsse auf die verantwortlichen Enzyme und Stoffwechselwege zulässt.
Eine besondere Herausforderung liegt darin, dass das Metabolom aus sehr unterschiedlichen Verbindungen besteht und zudem starke Konzentrationsunterschiede vorliegen. Die heute routinemäßig für Metabolomics verwendeten analytischen Techniken basieren auf Kernspinresonanzspektroskopie (NMR) und Massenspektrometrie (MS).
Die NMR-Spektroskopie wird hauptsächlich für die Identifizierung und Quantifizierung von kleinen, polaren Molekülen eingesetzt. Es wird keine oder nur eine minimale Probenvorbereitung benötigt, die Methode ist direkt quantitativ und die Proben werden durch die Messung nicht zerstört. Der größte Nachteil liegt in der relativ geringen Empfindlichkeit dieser Methode, sodass niedrig konzentrierte Metabolite nicht erfasst werden können. Die Vorteile MS-basierter Methoden sind die hohe Empfindlichkeit, die hohe Selektivität und die Möglichkeit zur Kopplung mit chromatographischen Methoden wie Gaschromatographie (GC) oder Flüssigkeitschromatographie (LC).
Sie sind daher ideal zur Analyse von organischen Verbindungen in komplexen Matrizes. Es gibt verschiedene methodische Ansätze im Bereich Metablomics wie das Metabolic Fingerprinting, Metabolic Profiling und Targeted Metabolomics. Beim Metabolic Fingerprinting handelt es sich um eine Hochdurchsatz-Gesamtanalyse des Metaboloms, um ohne Identifizierung einzelner Verbindungen zwischen Proben unterschiedlicher Herkunft oder unterschiedlichem biologischen Status zu differenzieren. Nach einer aufwendigen Datenaufbereitung werden die gewonnenen Daten anschließend verglichen, um Unterschiede zwischen verschiedenen Kohorten zu finden. Dies ist sicher ein guter Ansatz, um neue Biomarker zu finden, eine funktionelle Interpretation der Daten ist aber nicht möglich.
Dazu muss man die für die beobachteten Unterschiede relevanten Metabolite erst identifizieren und anschließend quantifizieren.
Beim Metabolic Profiling fokussiert man auf eine spezielle Gruppe von Metaboliten, die mit einem bestimmten Stoffwechselweg assoziiert ist. Bei Targeted Metabolomics handelt es sich um quantitative Bestimmung einiger Zielanalyten. Der Übergang zwischen diesen Ansätzen ist fließend wobei generell gilt, dass sich die Qualität der Daten für den einzelnen Analyten durch die Fokussierung auf eine geringere Anzahl von Analyten verbessert. Beide Methoden erlauben eine direkte biochemische Interpretation der Daten.
Es konnten verschiedene für chronische Nierenerkrankungen relevante Metabolite identifiziert werden. Darunter zum Beispiel Verbindungen aus dem Dimethylarginin-Metabolismus oder Intermediate des Tryptophan-Abbaus.
So findet man eine Erhöhung des Plasma-SDMA-Spiegels und Zunahme der Tryptophan-Abbauprodukte im Serum mit Fortschreiten der Nierenerkrankung. Solche Marker sind wichtig, um die Pathomechanismen hinter den Nierenschäden aufzuklären, das Fortschreiten der CKD zu beurteilen und eine geeignete Behandlung auszuwählen. Eine vielversprechende Anwendung von Targeted Metabolomics ist die Identifizierung von Biomarkern als Indikatoren der Nierenfunktion. In der klinischen Praxis verwendet man Serum-Kreatinin und die geschätzte glomeruläre Filtrationsrate (eGFR), die bekanntermaßen relativ unempfindlich und eigentlich Marker für späte CKD-Stadien sind. Mehreren Studien zeigen, dass die Plasma-SDMA-Konzentration gut mit der eGFR korreliert. Die Kombination mit anderen Metaboliten zu multiparametrischen Biomarkern, die die individuelle biologische Variabilität kompensieren, könnte helfen, Patienten bereits in frühen Stadien der chronischen Nierenerkrankung mit hoher diagnostischer Qualität zu identifizieren.

Ursprünglich erschienen:
Neph 01|2012
Neph 01|2012