


























































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Integrative Analyse von Omics-Daten zu chronischen Nierenerkrankungen
30. März 2012
Omics-Datensätze in der nephrologischen Forschung
Auf Ebene des Genoms (Genomics) werden Punktmutationen, sogenannte single nucleotide polymorphisms (SNP), entweder mittels Sequenzierung, Real-time-PCR oder arraybasierter Systeme ermittelt. Diese Mutationen sind Abweichungen in der Basensequenz eines Gens, die sich in weiterer Folge im Phänotyp auswirken und unter anderem ein erhöhtes Risiko für verschiedene Krankheiten darstellen. Da SNP im Genom sehr häufig vorkommen, sind große Patientenzahlen notwendig, um signifikante Assoziationen zwischen SNP und einer Erkrankung zu identifizieren. In einer Studie von Köttgen wurden SNP ermittelt, die mit chronischen Nierenerkrankungen assoziiert sind (Köttgen et al., 2010 Nat Genet). Eines der am stärksten betroffenen Gene war Uromodulin, auch bekannt als Tamm-Horsfall-Protein, das auch im Harn nachgewiesen werden kann. Allerdings konnte selbst mit einer Kombination von mehreren identifizierten SNP nur ein geringer Prozentsatz der Varianz der glomerulären Filtrationsrate (GFR) erklärt werden, was nahelegt, dass es entweder noch weitere derzeit nicht entdeckte Genorte geben muss, die mit einer reduzierten GFR in Verbindung stehen, oder dass Umweltfaktoren eine wesentlich größere Rolle spielen als die genetische Prädisposition.
 In Transcriptomics-Studien werden Unterschiede auf Ebene der mRNA ermittelt. Eine der ersten Pilotstudien auf diesem Gebiet stammt aus dem Jahr 2004 von Baelde und Kollegen, die Expressionmuster bei Patienten mit und ohne diabetischer Nephropathie an einem kleinen Patientenkollektiv verglichen haben (Baelde et al., 2004 Am J Kidney Dis). RNA wurde hierbei aus Glomeruli von Patienten isoliert und die Konzentrationen mit Hilfe von Mikroarrays verglichen. Nach einer statistischen Analyse blieben von den gesamt 12.000 untersuchten Genen 615 übrig, welche differenziell zwischen Patienten mit diabetischer Nephropathie und gesunden Probanden reguliert waren. 519 Gene zeigten bei Patienten mit diabetischer Nephropathie eine verminderte und 96 eine erhöhte Genexpression. Die Autoren verifizierten ihre Ergebnisse stichprobenartig mit einer weiteren Technologie, der Realtime-PCR (Polymerasekettenreaktion), die zwar den Goldstandard in der Transcriptomics darstellt, aber den Nachteil hat, dass für jedes zu untersuchende Sequenzstück ein eigenes Experiment gestartet werden muss. Die kontrollierten Gene waren Nephrin, VEGF und TGF-ß, die eine sehr gute Übereinstimmung mit den Mikroarraydaten zeigten. Seit dieser Pilotstudie wurde eine Reihe weiterer Transcriptomics-Studien sowohl zu diabetischer Nephropathie wie auch zu anderen Formen der chronischen Nierenerkrankung durchgeführt (Rudnicki et al., 2007 Kidney Int; Rudnicki et al., 2009 Lab Invest).
In Transcriptomics-Studien werden Unterschiede auf Ebene der mRNA ermittelt. Eine der ersten Pilotstudien auf diesem Gebiet stammt aus dem Jahr 2004 von Baelde und Kollegen, die Expressionmuster bei Patienten mit und ohne diabetischer Nephropathie an einem kleinen Patientenkollektiv verglichen haben (Baelde et al., 2004 Am J Kidney Dis). RNA wurde hierbei aus Glomeruli von Patienten isoliert und die Konzentrationen mit Hilfe von Mikroarrays verglichen. Nach einer statistischen Analyse blieben von den gesamt 12.000 untersuchten Genen 615 übrig, welche differenziell zwischen Patienten mit diabetischer Nephropathie und gesunden Probanden reguliert waren. 519 Gene zeigten bei Patienten mit diabetischer Nephropathie eine verminderte und 96 eine erhöhte Genexpression. Die Autoren verifizierten ihre Ergebnisse stichprobenartig mit einer weiteren Technologie, der Realtime-PCR (Polymerasekettenreaktion), die zwar den Goldstandard in der Transcriptomics darstellt, aber den Nachteil hat, dass für jedes zu untersuchende Sequenzstück ein eigenes Experiment gestartet werden muss. Die kontrollierten Gene waren Nephrin, VEGF und TGF-ß, die eine sehr gute Übereinstimmung mit den Mikroarraydaten zeigten. Seit dieser Pilotstudie wurde eine Reihe weiterer Transcriptomics-Studien sowohl zu diabetischer Nephropathie wie auch zu anderen Formen der chronischen Nierenerkrankung durchgeführt (Rudnicki et al., 2007 Kidney Int; Rudnicki et al., 2009 Lab Invest).
Während Transcriptomics-Profile vorwiegend aus Gewebeschnitten abgeleitet wurden, sind die meisten Proteomics-Studien entweder an Plasma oder Harn durchgeführt worden. Beispielhaft hierfür ist die Studie von Otu und Kollegen, deren Fokus auf dem Vergleich von Proteinmustern im Urin von Pima-Indianern lag (Otu et al., 2007 Diabetes Care). Durch die Bestimmung von 28 Proteinen konnte die Kohorte von Pima-Indianern mit diabetischer Nephropathie von den Gesunden unterschieden werden. Durch weitere Analysen konnte das initiale Proteinpanel auf 12 Proteine reduziert werden, mit deren Messung in einer Validierungskohorte eine Sensitivität von 93 % und eine Spezifitiät von 86 % erreicht werden konnte.
In der Arbeit von Zhang wurden Blutproben (Seren) untersucht, um für die diabetische Nephropathie rele vante Metaboliten zu identifizieren (Zhang et al., Anal Chim Acta 2009). Um die komplexe Matrix im Serum aufzutrennen, wurde die Ultraperformance-Liquid-Chromatographie (UPLC) in Verbindung mit der Massenspektroskopie (MS) verwendet, um die aufgetrennten Metaboliten zu identifizieren. Unter den identifizierten Metaboliten waren unter anderem Substanzen wie Dihydrosphingosine, Phytosphingosin und Leucin zu finden, Moleküle, die im Aminosäure- und Phospholipidstoffwechsel eine Rolle spielen.
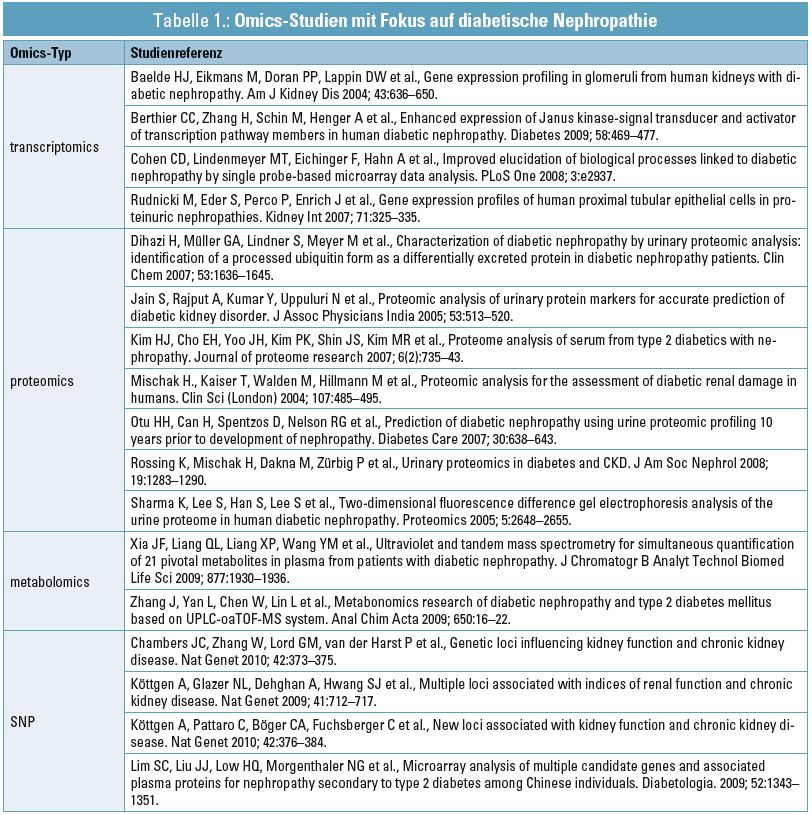
Tabelle 1 gibt einen Überblick über Omics-Studien mit Fokus auf der diabetischen Nephropathie. Die bisherigen Studien waren hauptsächlich auf einen Typ von Omics-Daten fokussiert, und nur sehr wenige Studien integrierten Omics-Daten verschiedenen Typs. Das folgende Kapitel soll einige Analysestrategien aufzeigen, die ein Verschneiden verschiedener Omics-Datensätze ermöglichen.
Analysestrategien
Fragen, die sich aus den einzelnen Omics-Studien ergeben, sind unter anderem, inwieweit sich eine Mutation auf Genebene auf die Expression des jeweiligen Gens auswirkt und danach auf die Konzentration (oder Funktion) des zu kodierenden Proteins. Die singulären Resultate aus den Omics-Studien können nun auf verschiedenen Ebenen verglichen werden, nämlich
- auf Ebene der direkten Schnittmenge,
- auf Ebene angereicherter funktionaler Kategorien,
- auf Ebene von definierten molekularen Pathways, oder
- auf Ebene von Protein-Protein-Interaktionsnetzwerken.
Diese vier Ebenen sind in Abbildung 1 veranschaulicht und werden in den weiteren Kapiteln näher erläutert.
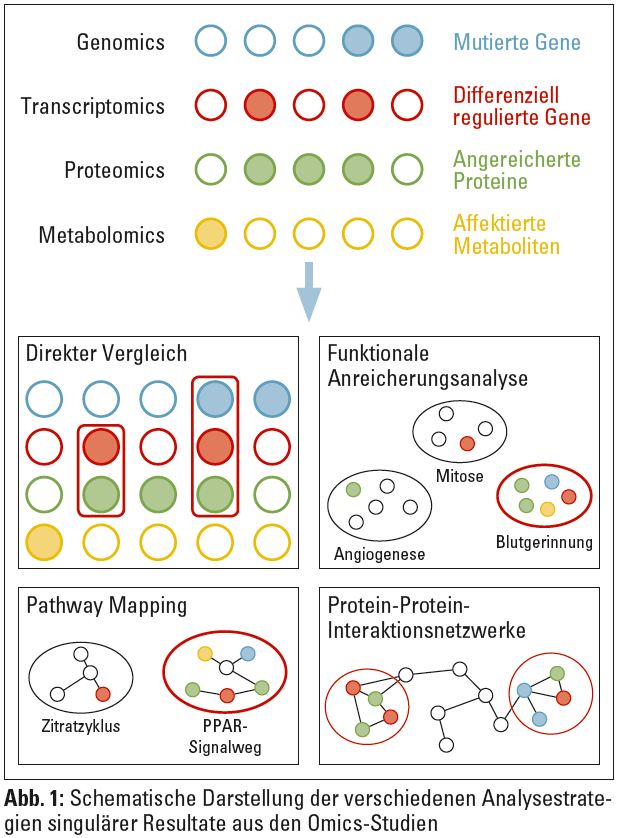 Direkter Vergleich von molekularen Entitäten: Der direkte Vergleich von als relevant erachteten molekularen Entitäten ist für SNP, Transkripte und Proteine durch ein Mappen auf das entsprechende Gen realisierbar. Hierfür gibt es eine Reihe von Annotationsdatenbanken, wie zum Beispiel GeneCards (http://www.genecards.org) oder die Datenbanken des National Center for Biotechnological Information (NCBI; http://www.ncbi.nlm.nih.gov).
Direkter Vergleich von molekularen Entitäten: Der direkte Vergleich von als relevant erachteten molekularen Entitäten ist für SNP, Transkripte und Proteine durch ein Mappen auf das entsprechende Gen realisierbar. Hierfür gibt es eine Reihe von Annotationsdatenbanken, wie zum Beispiel GeneCards (http://www.genecards.org) oder die Datenbanken des National Center for Biotechnological Information (NCBI; http://www.ncbi.nlm.nih.gov).
Metabolite können nicht direkt mit Genen verglichen werden, diese können jedoch zu Proteinen gelinkt werden, die an deren Abbau oder deren Prozessierung beteiligt sind. Verbindungen zwischen Metaboliten und deren assoziierten Proteinen sind zum Beispiel über die Human Metabolome Database (http://www.hmdb.ca) verfügbar. Molekulare Entitäten, die auf verschiedenen molekularen Ebenen gefunden worden sind, werden als relevanter erachtet.
 Funktionale Anreicherungsanalyse: Bei der funktionalen Anreicherungsanalyse werden die einzelnen molekularen Entitäten zu funktionalen Kategorien gelinkt, soweit bereits aus vorangegangenen Studien bekannt. Eine der am häufigsten verwendeten Datenquellen mit funktionaler Annotation zu Genen und Proteinen wurde vom Gene Ontology Consortium (www.geneontology.org) entwickelt. Diese Ontologie ist mit den drei Hauptkategorien „biological process“, „molecular function“ und „cellular component“ hierarchisch aufgebaut.
Funktionale Anreicherungsanalyse: Bei der funktionalen Anreicherungsanalyse werden die einzelnen molekularen Entitäten zu funktionalen Kategorien gelinkt, soweit bereits aus vorangegangenen Studien bekannt. Eine der am häufigsten verwendeten Datenquellen mit funktionaler Annotation zu Genen und Proteinen wurde vom Gene Ontology Consortium (www.geneontology.org) entwickelt. Diese Ontologie ist mit den drei Hauptkategorien „biological process“, „molecular function“ und „cellular component“ hierarchisch aufgebaut.
Abbildung 2 stellt einen Auszug aus der Gene Ontology dar mit der hervorgehobenen Kategorie „complement activation“. Dieser Kategorie sind 194 humane Gene zugeordnet. Mit einer Liste an molekularen Entitäten, dem üblichen Ergebnis eines Omics-Experiments, lassen sich durch die Zuweisung auf die funktionalen Kategorien nun jene Kategorien identifizieren, die statistisch signifikant angereichert und so als relevant zu erachten sind. Hierfür wird üblicherweise ein Chi-Quadrat-Test eingesetzt.
Auf dem Gebiet der Nephrologie hat sich auch ein Konsortium, die Renal Gene Ontology Initiative (http://www.geneontology.org/GO.renal.shtml) gebildet, mit dem Ziel, die Annotation nierenrelevanter Prozesse zu verbessern. Zum einen soll die Anzahl an Kategorien in der Ontologie erweitert werden und zum anderen soll die Zuordnung nierenspezifischer Gene, respektive Proteine verbessert werden.
Pathway Mapping: Für Schlüsselprozesse im Stoffwechsel sind die Verbindungen von Proteinen und Metaboliten bekannt und in Pathway-Datenbanken wie zum Beispiel KEGG (http://www.genome.jp/kegg), PANTHER (http://www.pantherdb.org) oder Reactome (http://www.reactome.org) gesammelt. Diese Datenbanken können in einer ähnlichen Art und Weise wie die funktionellen Kategorien herangezogen werden, um stark affektierte Pathways anhand von bekannten molekularen Entitäten zu identifizieren. Pathway-Datenbanken halten allerdings nur einen Teil aller bekannten Proteine. Wesentlich mehr Information ist in Protein-Protein-Interaktionsdatenbanken enthalten, die zusätzlich Information über Zusammenhänge geben können, wie unten näher beschrieben.
Protein-Protein-Interaktionsnetzwerke: Ausgehend von Informationen zu Protein-Protein-Interaktionen können Interaktionsnetzwerke erstellt und in weiterer Folge jene Bereiche des Netzwerks identifiziert werden, die anhand einer Liste an relevanten molekularen Entitäten affektiert sind. Zu den häufig eingesetzten Protein-Protein-Interaktionsdatenbanken gehören IntAct (http://www.ebi.ac.uk/intact), BioGrid (http://thebiogrid.org) oder auch die bereits oben erwähnte Reactome-Datenbank.
Cross-Omics-Studie zu diabetischer Nephropathie
Mitarbeiter des SysKid-Teams veröffentlichten kürzlich Ergebnisse zur Verschneidung einer Reihe von Omics-Datensätze die diabetische Nephropathie betreffend (Fechete et al., Proteomics Clin Appl 2010). Hierin wurden die Ergebnisse von 17 Einzelstudien inkludiert: je 4 Transkriptomics-und SNP-Studien, 2 Metabolomics-Studien und 7 Proteomics-Studien (Tab. 1).
 In Summe konnten wir 1.059 molekulare Entitäten aus den Studien extrahieren, wobei der Großteil aus Transkriptomics-Studien kam.
In Summe konnten wir 1.059 molekulare Entitäten aus den Studien extrahieren, wobei der Großteil aus Transkriptomics-Studien kam.
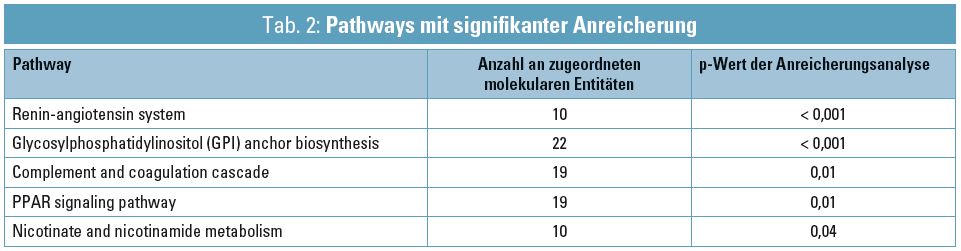
Tabelle 2 gibt einen Überblick über jene Pathways, die basierend auf dem Satz an molekularen Entitäten als signifikant angereichert identifiziert wurden. Für die nephrologische Therapie und Diagnostik relevante Moleküle aus diesen Pathways sind unter anderem derzeit Schwerpunkt des Forschungsprojekts SysKid (Systems Biology towards Novel Chronic Kidney Disease Diagnosis and Treatment). Der Fokus liegt auf dem frühen Stadium der chronischen Nierenerkrankung, hauptsächlich ausgelöst durch Diabetes oder Bluthochdruck.
NEPHROSpot
Resultate aus Omics-Studien in der Nephrologie haben in den letzten Jahren zum weiteren Verständnis des Krankheitsverlaufs geführt und eine Reihe an potenziellen Biomarkern hervorgebracht, die derzeit bereits zum Teil in Validierungsstudien für den klinischen Einsatz getestet werden. Die bioinformatische Auswertung und Verschneidung der Resultate spielt hierbei eine maßgebliche Rolle in der Selektion der Markerproteine und der Aufklärung der molekularen Prozesse im Zuge der Krankheitsentstehung und deren Verlauf.

Ursprünglich erschienen:
Neph 01|2012
Neph 01|2012