


























































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Lupusnephritis 2012
31. Dezember 2012
Die Nierenbeteiligung stellt die häufigste schwere Manifestation des systemischen Lupus erythematodes (SLE) dar. Zum Zeitpunkt der klinischen Erstmanifestation weisen bereits bis zu 50 % der Patienten eine renale Beteiligung auf. Dieser Prozentsatz steigt im Follow-up auf bis zu 60 % an und korreliert nicht zwangsläufig mit weiteren Organmanifestationen. Jeder Hinweis auf eine renale Beteiligung stellt die Indikation zur diagnostischen Nierenbiopsie dar, zumal klinische Parameter nicht ausreichend mit der Nierenhistologie korrelieren. Die spezifische Therapie basiert auf der Nierenhistologie, die rechtzeitige (frühe!) Etablierung einer adäquaten Therapie ist für die Morbidität und Mortalität dieser Patienten entscheidend. Etwa 10–30 % der Patienten mit Lupusnephritis (LN) werden trotz Therapie innerhalb von 15 Jahren dialysepflichtig. Generell verläuft die LN bei Kindern und Jugendlichen häufig schwerer als bei Erwachsenen.
Wann ist eine Nierenbiopsie indiziert?
Erste Hinweise für eine Nierenbeteiligung gibt das Harnsediment. Dies sollte bei Patienten mit SLE regelmäßig überprüft werden. Die Indikation für eine Nierenbiopsie wird beim SLE strenger als bei anderen Erkrankungen mit Nierenbeteiligung gestellt, da bereits geringe Veränderungen im Harnsediment bei auch (noch) normaler Nierenfunktion Ausdruck einer therapiebedürftigen LN sein können und daher nicht nur diagnostisch, sondern auch prognostisch relevant sind. Nach den EULAR/ERA-EDTA-Empfehlungen 2012 stellen eine glomeruläre Mikrohämaturie und/oder eine reproduzierbare Proteinurie von > 0,5 g Eiweiß/24 h auch bei (noch) normaler Nierenfunktion eine Indikation zur Nierenbiopsie dar. Ebenso sollte eine ungeklärte Leukozyturie (nach Ausschluss einer Infektion) in einzelnen Fällen bioptisch abgeklärt werden. Liegt eine eingeschränkte Nierenfunktion bei unauffälligem Sediment vor, so entscheiden Dynamik und Anamnese über das weitere Vorgehen. Wichtig ist es auch, das mögliche Vorliegen von Antiphospholipid-Antikörpern oder Lupus-Antikoagulans zu überprüfen.
Klinik und Histologie der Lupusnephritis
Die LN weist das typische Bild einer Immunkomplex-Glomerulonephritis auf. In der Immunhistologie (oder Immunfluoreszenz) findet sich ein positives Staining für IgA, IgG, IgM sowie Komplementfaktoren („full house“). Es gibt verschiedene histologische Typen der Lupusnephritis, die sich hinsichtlich Prognose und therapeutischem Procedere unterscheiden.
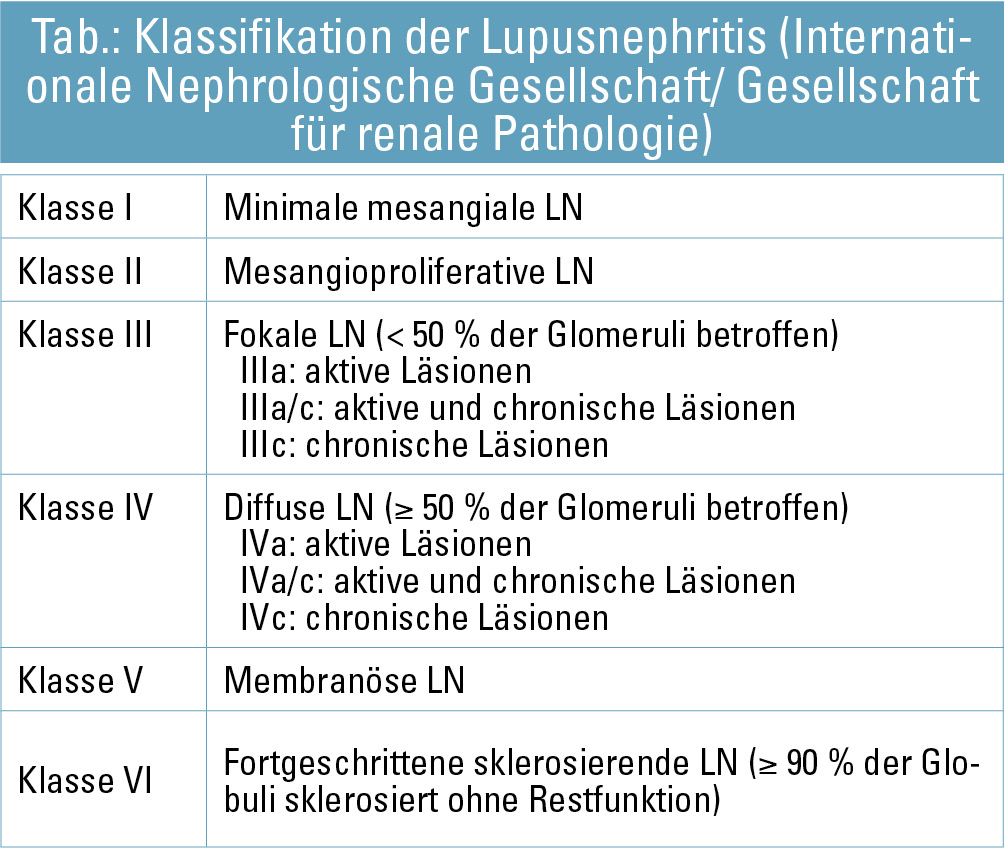
Die Evaluierung der Nierenbiopsie sollte daher nach dem ISN/RPS-2003-Klassifikationssystem erfolgen, welches sich primär auf glomeruläre Veränderungen bezieht (Tab.). Darüber hinaus sind auch die Beurteilung tubulointerstitieller und vaskulärer Veränderungen sowie der Aktivitätsindex und der Chronizitätsindex für das therapeutische Vorgehen entscheidend.
Die LN Class I ist charakterisiert durch lichtmikroskopisch unauffällige Glomeruli sowie geringe Immunglobulinablagerungen und bedarf aufgrund der guten renalen Prognose keiner speziellen Therapie.
Die LN Class II ist definiert durch eine mesangioproliferative Glomerulonephritis mit mesangialen Immunkomplex(IC)-Ablagerungen ohne lichtmikroskopische Veränderungen. Klinisch findet sich meist eine Mikrohämaturie und/oder auch eine geringe Proteinurie (meist < 1 g/24 h). Ein Übergang in eine aggressivere Form der LN ist möglich.
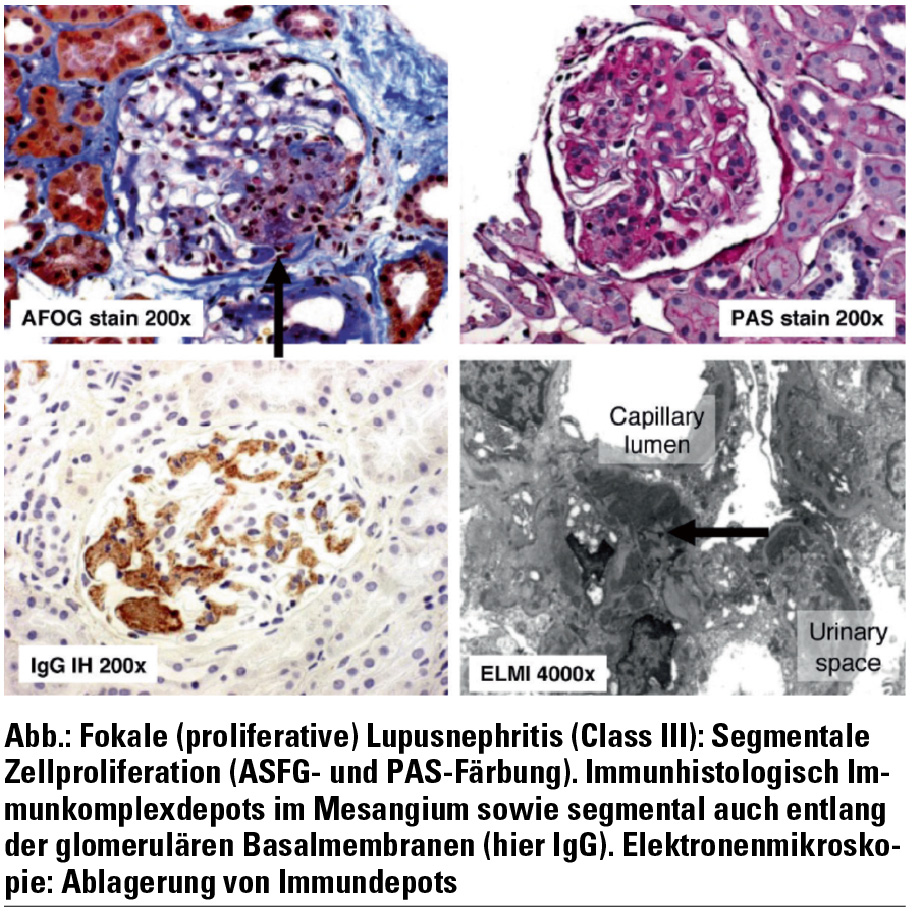
Die fokal-segmentale proliferative Glomerulonephritis (LN Class III, Abb.) ist neben mesangialen Veränderungen durch segmentale intrakapilläre Proliferationen und mitunter auch nekrotische Veränderungen charakterisiert. Im Unterschied zur LN Class IV sind weniger als 50 % der Glomeruli betroffen. Die renale Prognose ist variabel. Sind weniger als 20 % der Glomeruli betroffen, die Nierenfunktion normal und liegt kein nephrotisches Syndrom vor, so liegt die renale 5-Jahres-Überlebensrate bei etwa 95 %. Prognostisch ungünstige Faktoren sind hingegen zelluläre Proliferationen, Nekrosen und ausgedehnte subendotheliale IC‑Ablagerungen in mehr als 40 % der Glomeruli sowie klinisch das Vorliegen eines nephrotischen Syndroms.
Die häufigste und schwerste Form stellt die diffus-proliferative Glomerulonephritis (LN Class IV) dar, welche histologisch der LN Class III gleicht, es sind jedoch per definitionem mehr als 50 % der Glomeruli betroffen. Darüber hinaus finden sich häufig zusätzliche Veränderungen wie ausgeprägte IC-Ablagerungen in den glomerulären Kapillarwänden unter dem lichtmikroskopischen Bild einer membranoproliferativen Glomerulonephritis sowie prognostisch ungünstige extrakapilläre proliferative Veränderungen (zelluläre Halbmonde, „crescents“). Der Verlauf ist aggressiv, die Patienten weisen oft schon zum Zeitpunkt der Diagnosestellung eine eingeschränkte Nierenfunktion auf. Leitsymptome sind eine (akzelerierte) arterielle Hypertonie, eine ausgeprägte Proteinurie oder ein nephrotisches Syndrom, obligat eine (Mikro-)Hämaturie. Die renale 5-Jahres-Überlebensrate wird in der Literatur mit 50–80 % angeben.
Die LN Class V entspricht dem Bild einer membranösen Glomerulonephritis und ist durch eine Verdickung der Kapillarwand charakterisiert. Eine mesangiale Proliferation ist meist nur gering ausgeprägt, die Immunhistologie („full house“) ist hilfreich in der differenzialdiagnostischen Abgrenzung zur idiopathischen membranösen Glomerulonephritis. Klinischer Leitbefund ist die Proteinurie bis zum nephrotischen Syndrom. Die Prognose ist deutlich besser als die der proliferativen Formen. Es soll jedoch darauf aufmerksam gemacht werden, dass in vielen Fällen auch ein Mischbild von LN Class III/V oder LN Class IV/V vorliegt. Das Bild der LN Class VI entspricht mit einer globalen Sklerose von mehr als 90 % der Glomeruli jenem einer „end stage kidney“.
Therapie der Lupusnephritis
Die Therapieentscheidung bei Lupusnephritis basiert auf der diagnostischen Nierenbiopsie. Hauptziel der Behandlung ist die Langzeiterhaltung der Nierenfunktion bei möglichst geringer Toxizität und gleichzeitiger Relaps-Prävention. Initiales Ziel ist eine komplette renale Remission, definiert als Proteinurie < 0,5 g/24 h bei normaler oder stabiler Nierenfunktion. Von einem Therapierespons spricht man bei einer zumindest 50%igen Reduktion der Proteinurie und einer Normalisierung oder Stabilisierung der Nierenfunktion. Dies sollte etwa 6–12 Monate nach Initialisierung der Therapie erreicht sein. Eine volle Remission wird bei LN oft spät erreicht, mitunter erst nach 2 Jahren.
Die LN Class I und II bedarf keiner spezifischen immunsuppressiven Therapie, jedoch einer regelmäßigen nephrologischen Kontrolle, da diese Formen jederzeit in eine andere histologische Form übergehen können. Hier stehen ACE-I und/oder ARB im Vordergrund. Persistiert bei LN Class II trotz ACE-I/ARB-Therapie eine Proteinurie von > 1 g/24 h (was bei LN II selten ist), so kann in dieser Situation auch eine spezifische Therapie diskutiert werden. Aufgrund der hohen Chronizität der LN Class VI ist kein Benefit einer Immunsuppression zu erwarten, und die Vorbereitung zur Nierenersatztherapie steht im Vordergrund.
Die proliferativen Formen der LN, dazu gehören LN Class III und IV, sind die schwersten Verlaufsformen. Für die meisten Patienten mit LN Class IIIA oder IIIA/C (± V) und Class IVA oder IVA/C (± V) ist eine Induktionstherapie mit Mykophenolat-Mofetil (MMF) 3 g/Tag oder Low-Dose-Zyklophosphamid i. v. (EURO-Lupus-Schema: 0,5 g 6-mal alle 14 Tage) in Kombination mit oralen Kortikosteroiden (initial 3-mal Bolus von Methylprednisolon 500–750 mg i. v., gefolgt von 0,5 mg/kg/Tag für 4 Wochen, dann langsam Reduktion nach klinischer Symptomatik) empfohlen. Ungünstige prognostische Faktoren in der Nierenhistologie (z. B. zelluläre Halbmonde) oder eine rasche Verringerung der GFR können den Einsatz von höher dosiertem Zyklophosphamid (Zyklophosphamid i. v. 0,75–1g/m2 für 6 Monate (7 Boli) sowie Plasmaseparation (oder Immunadsorption) erforderlich machen. Für schwere oder therapierefraktäre Patienten stellt Rituximab eine sehr gute Alternative dar.
Zu erwähnen ist, dass in den meisten großen klinischen Studien Patienten mit raschem Abfall der GFR und rapidem Verlauf der LN ausgeschlossen waren, wenngleich sie die Basis für die Guidelines darstellen. In einem rezenten Review wurde bei Patienten mit > 15 % „crescents“ und eingeschränkter Nierenfunktion ein möglicher Benefit von Zyklophosphamid im Vergleich zu MMF betreffend Relaps und ESRD diskutiert, dies bei gleichem Erfolg betreffend die initiale Remission.
Milde Formen einer reinen LN Class V bedürfen primär einer Therapie mit ACE-I und/oder ARB. Liegt eine Proteinurie von > 1 g trotz Ausreizung einer Therapie mit ACE-I/ARB vor, so stellt MMF (Zieldosis 3 g/Tag) in Kombination mit oralen Kortikosteroiden (0,5 mg/kg/Tag) als Initialtherapie die erste Wahl dar. Zyklophosphamid i. v., Kalzineurininhibitoren (Zyklosporin A, Tacrolimus) oder Rituximab sind weitere Alternativen.
Nach Ansprechen auf die Initialtherapie, definiert als eine zumindest 50%ige Reduktion der Proteinurie sowie Ausgangsnierenfunktion, kann nach 6–12 Monaten eine Remissionserhaltung mit MMF (2 g/Tag) oder Azathioprin (2 mg/kg/Tag) für mindestens 3 Jahre angestrebt und dann ausgeschlichen werden.
Die Begleittherapie bei Patienten mit Lupusnephritis umfasst die optimale Blutdruckeinstellung (120/75 mmHg), bevorzugt mit ACE-I ± ARB sowie die Kontrolle der Dyslipidämie mit Statinen. Hydroxychloroquin sollte bei allen Patienten im Hinblick auf eine Reduktion renaler Relapse sowie zur Reduktion des bei diesen Patienten ohnehin erhöhten kardiovaskulären Risikos Bestandteil der Therapie sein. Unter dieser im Vergleich nebenwirkungsarmen Substanz werden ein günstiger Effekt auf das Lipid- und Blutzuckerprofil, die Thrombogenität sowie die Inzidenz maligner Erkrankungen beobachtet. Weitere Therapien umfassen die orale Antikoagulation bei nephrotischem Syndrom bei Serumalbumin < 20 g/l (insbesondere bei positiven Antiphospholipid-Antikörpern!) sowie eine Osteoporose-Prophylaxe.
B-Zell-Depletion bei Lupusnephritis
Im Wesentlichen gibt es nur eine große randomisierte Studie zu Rituximab bei proliferativem LN (LUNAR-Studie). Nach einem Follow-up von 52 Wochen konnte kein Effekt einer zusätzlichen Applikation von Rituximab zur Standardtherapie mit MMF 3 g/Tag und hoch dosierten Kortikosteroiden auf die Remissionsrate beobachtet werden. In der Rituximab-Gruppe fand sich jedoch ein signifikanter Abfall der dsDNA-AK, ein Anstieg von C3 und C4 und – besonders wichtig – keine Zunahme der Nebenwirkungen. Diskutiert wird, dass unter Rituximab mehr Patienten eine partielle Remission (wenngleich n. s.) erreichten, die Studie jedoch konzipiert wurde, um eine komplette Remission statistisch zu erfassen, was bei LN oft länger dauern kann. Das kurze Follow-up der Studie wird auch im Hinblick auf den renalen Respons, der unter Rituximab später eintreten kann, kritisiert. Das „Add on“-Design von Rituximab in Kombination mit der Standardtherapie könnte ebenfalls den Benefit maskieren. Aus einem rezenten Review geht hervor, dass in 21 Studien ca. 300 Patienten Rituximab meist für relapsierende oder therapierefraktäre LN erhielten, oft in Kombination mit CYC oder MMF. In Studien mit einem Follow-up von > 1 Jahr sprach ein Drittel der Patienten mit refraktärer/relapsierender LN auf Rituximab an. Es wurde jedoch kein Benefit einer Kombination mit anderen Immunsuppressiva (CYC, MMF) gefunden.
Monitoring
Zur Evaluierung des Therapieerfolges und etwaiger Nebenwirkungen sind in den ersten 3 Monaten ambulante Begutachtungen im Abstand von 2 bis 4 Wochen erforderlich. Die Intervalle können je nach Therapieerfolg entsprechend verlängert werden. In jedem Falle sind lebenslange Kontrollen notwendig. Dabei muss auch die extrarenale Krankheitsaktivität überprüft werden. Für das nephrologische Follow-up sind die Bestimmung von Serumkreatinin (GFR), Proteinurie, Harnsediment (Mikrohämaturie, Zylinder, Leukurie), Serum C3, Anti-ds-DANN-Titer, Blutbild und Serumalbumin die Basis.
Eine Rebiopsie sollte bei therapierefraktären Patienten sowie bei Verdacht auf Relapse durchgeführt werden, da neben einer Progression auch der Switch zu einer anderen histologischen LN-Class möglich ist. Ein schleichender Kreatininanstieg stellt eine weitere Indikation dar, da neben Veränderungen der Aktivität vor allem die Chronizität für die renale Prognose entscheidend ist, und eine Zunahme der Chronizität Ausdruck einer Unterbehandlung sein kann. Der Verdacht auf einen Relaps besteht bei einem (reproduzierbaren) Kreatininanstieg von > 30 % (oder Abfall der GFR um > 10 %) bei Vorliegen eines „aktiven“ Harnsediments, also Hämaturie > 10 Erys (unabhängig von der Proteinurie) und/oder Neuauftreten bzw. Anstieg einer Proteinurie.
Sollte eine Nierenersatztherapie erforderlich werden, so kommen prinzipiell alle Formen in Frage. Es sollte jedoch das erhöhte Risiko für Shuntthrombosen bei Patienten mit Antiphospholipid-AK und das generell erhöhte Infektrisiko bedacht werden. Eine Nierentransplantation kann bei Patienten ohne oder mit einer nur sehr geringen Krankheitsaktivität über zumindest 3–6 Monate in Erwägung gezogen werden. Die Bestimmung von Antiphospholipid-AK ist in der Transplantationsvorbereitung von Bedeutung, da diese mit einem erhöhten Thromboserisiko einhergeht und in diesem Setting eine perioperative Antikoagulation diskutiert werden muss.

AutorIn: Univ.-Doz. Dr. Irmgard Neumann
6. Medizinische Abteilung mit Nephrologie und Dialyse, Wilhelminenspital Wien

Ursprünglich erschienen:
Neph 04|2012
Neph 04|2012