


























































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Membranöse Glomerulopathie
31. Dezember 2012
Die membranöse Nephropathie (MN) ist eine der häufigsten nephrotischen Erkrankungen im Erwachsenenalter bei nichtdiabetischen Patienten. Die Bezeichnung „membranös“ bezieht sich auf die Erscheinungsform in der Lichtmikroskopie, nämlich eine Verdickung der glomerulären Basalmembran. Man unterscheidet eine häufigere primäre (idiopathische) Form und eine seltenere sekundäre Form der MN. Bei beiden Formen finden sich subepitheliale Antigen-Antikörper-Komplexe ohne wesentliche bzw. nur mit schwacher systemischer Aktivierung des Komplementsystems. Bei der idiopathischen membranösen Nephropathie (IMN) ist das Antigen podozytären Ursprungs bzw. unbekannt. Im Gegensatz dazu wird die Immunkomplexbildung bzw. -ablagerung bei sekundärer MN auf Ursachen wie eine Hepatitis-B-Antigenämie, systemischen Lupus erythematodes, Thyreoiditis, Malignome oder auch Medikamente (Gold, Penicillamin, NSAR u. a.) zurückgeführt. Insbesondere bei der IMN ist in den vergangenen Jahren durch die Entdeckung des podozytären Antigens Phospholipase-A2-Rezeptor (PLA2R) und den im Serum zirkulierenden PLA2R-Antikörpern ein entscheidender Schritt in der Aufklärung der Pathogenese und in der Diagnostik gelungen. Diese Entdeckungen und damit verbundene neue therapeutische Ansätze könnten die Standardtherapie, den Verlauf und die Rezidivneigung einer IMN nach Nierentransplantation grundlegend verändern.
Epidemiologie
MN ist vor allem bei über 40-jährigen Erwachsenen die häufigste Form eines nephrotischen Syndroms und verantwortlich für ca. 20 % aller Fälle. In einer Durchsicht der Nierenbiopsien, durchgeführt an der Nephrologie Innsbruck zwischen den Jahren 2000 und 2010, stellte die MN gemeinsam mit der Vaskulitis nach der IgA-Nephritis die zweithäufigste Biopsiediagnose überhaupt dar. Bei den über 60-jährigen Männern war sie sogar die häufigste Diagnose (nichtpublizierte Daten). Eine MN tritt bei allen Ethnien und bei beiden Geschlechtern auf. Bei der histologischen Diagnose einer MN bei einer jungen Frau sollte immer an eine Lupusnephritis gedacht werden. Eine MN ist bei Kindern eine seltene Diagnose. Meistens handelt es sich dann um eine sekundäre Form, assoziiert mit Hepatitis B oder einer Autoimmunerkrankung.
Pathogenese
In der vergangenen Dekade wurden im Verständnis der Pathogenese der MN große Fortschritte erzielt. Tierexperimentelle Studien, insbesondere das Modell der Heymann-Nephritis in der Ratte, führten zu dem Konzept des Podozytenrezeptors Megalin als Antigen für zirkulierende Antikörper. In Folge kommt es in situ zu einer Ablagerung von Antigen-Antikörper-Komplexen. Die subepithelial abgelagerten Immunkomplexe aktivieren lokal das Komplementsystem mit konsekutiver Ausbildung des „C5b-9 membrane attack complex“, der zu einer Hinaufregulation der Expression und der Translokation der NADPH-Oxidoreduktase an die Zelloberfläche führt. In weiterer Folge kommt es zu einer lokalen Überproduktion reaktiver Sauerstoffradikale, einer Schädigung der Podozytenmembran und ebenso auch der Proteine der glomerulären Basalmembran. Durch die Aktivierung intrazellulärer Signaltransduktionskaskaden werden das Aktin-Zytoskelett und die Integrität des Schlitzdiaphragmas gestört, was vermutlich für die Proteinurie von ausschlaggebender Bedeutung ist. Zusätzlich kommt es zu einer pathologischen Biosynthese von Kollagen Typ IV und Laminin, welches zu der typischen lichtmikroskopisch sichtbaren Verdickung der glomerulären Basalmembran führt. Obwohl es lokal zu einer Komplementaktivierung kommt, führt die Lokalisation der Immunkomplexe auf der subepithelialen, also der dem Blut abgewandten Seite zu keiner nennenswerten Aktivierung des systemischen Komplementsystems und der Leukozyten und somit zu keiner subendothelialen oder mesangialen Proliferation.
Humane Podozyten exprimieren kein Megalin, allerdings konnten mit dem Phospholipase-A2-Rezeptor (PLA2R) und der neutralen Endopeptidase (NEP) andere podozytäre Antigene identifiziert werden, die eine entscheidende Rolle bei der humanen MN spielen. In einer bahnbrechenden Arbeit von Beck und Kollegen (N Engl J Med 2009) konnte der M-Typ des PLA2R bei Patienten mit einer IMN als Antigen identifiziert werden. Dieses Protein ist ein Transmembranrezeptor, welcher von humanen Podozyten stark exprimiert wird. In dieser und in anderen Arbeiten konnten bei ca. 70% der Patienten mit einer IMN zirkulierende Antikörper gegen dieses Antigen nachgewiesen werden. Bei Patienten mit sekundären Formen einer MN (z. B. bei Lupusnephritis oder Hepatitis B), bei Patienten mit anderen nephrotischen Erkrankungen und bei gesunden Kontrollen waren bisher keine PLA2R-Antikörper nachweisbar. Interessanterweise handelt es sich bei diesen zirkulierenden Antikörpern um IgG4-Antikörper, also um jenen Subtyp, der histologisch bei einer IMN häufig gefunden wird. Zusätzlich konnte eine glomeruläre Kolokalisation des PLA2R und der IgG4-Immunkomplexe bei der IMN gezeigt werden, was die pathogenetische Relevanz dieser Antikörper und des Antigens noch mehr unterstreicht. Seit dem Jahr 2011 ist ein semiquantitativer indirekter Immunfluoreszenztest kommerziell erhältlich, der sowohl für den qualitativen Nachweis als auch für die Quantifizierung der PLA2R-Antikörper verwendet wird (siehe Diagnostik).
Auch genetische Faktoren scheinen bei der MN eine Rolle zu spielen. So konnten verschiedene Haplotypen (z. B. HLA-B8DR3), Polymorphismen (z. B. des Nephrin-Gens) und Genloci (z. B. 6p21 HLA-DQA1 und 2q24 PLA2R1) identifiziert werden, die mit einem höheren Risiko für das Auftreten einer MN oder der Wahrscheinlichkeit einer Remission/Progression der Erkrankung assoziiert waren.
Die NEP wird ebenso auf Podozyten exprimiert und ist das Antigen bei einer seltenen Form der antenatalen MN. Bei dieser Erkrankung haben die Mütter eine genetische Defizienz für das NEP-Protein und werden durch vorausgegangene Schwangerschaften durch das fetale NEP alloimmunisiert. Die Antikörper vom Typ IgG4 passieren die Plazentaschranke und führen zu einer MN beim Fetus und beim Neugeborenen mit subepithelialen Anti-NEP-NEP-Immunkomplexen. Nach dem Abbau der mütterlichen Anti-NEP-Antikörper kommt es zu einer spontanen Remission des nephrotischen Syndroms beim Säugling. Zu anderen seltenen Antigenen bei der IMN zählen bovines Serumalbumin, Aldosereduktase, Superoxid-Dismutase-2 und Alpha-Enolase.
Diagnostik
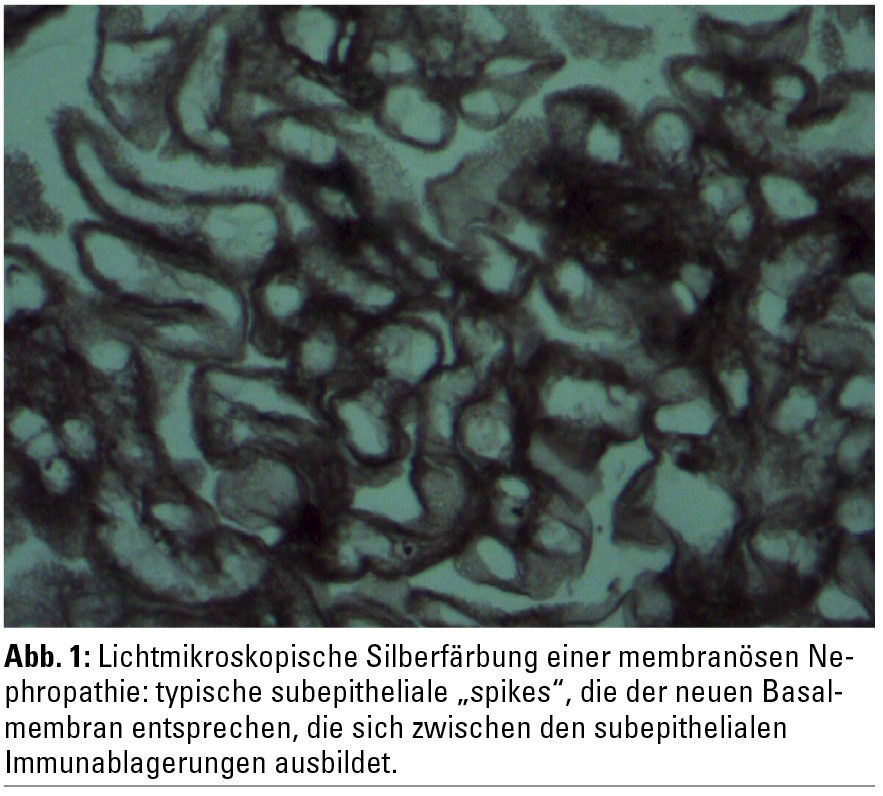
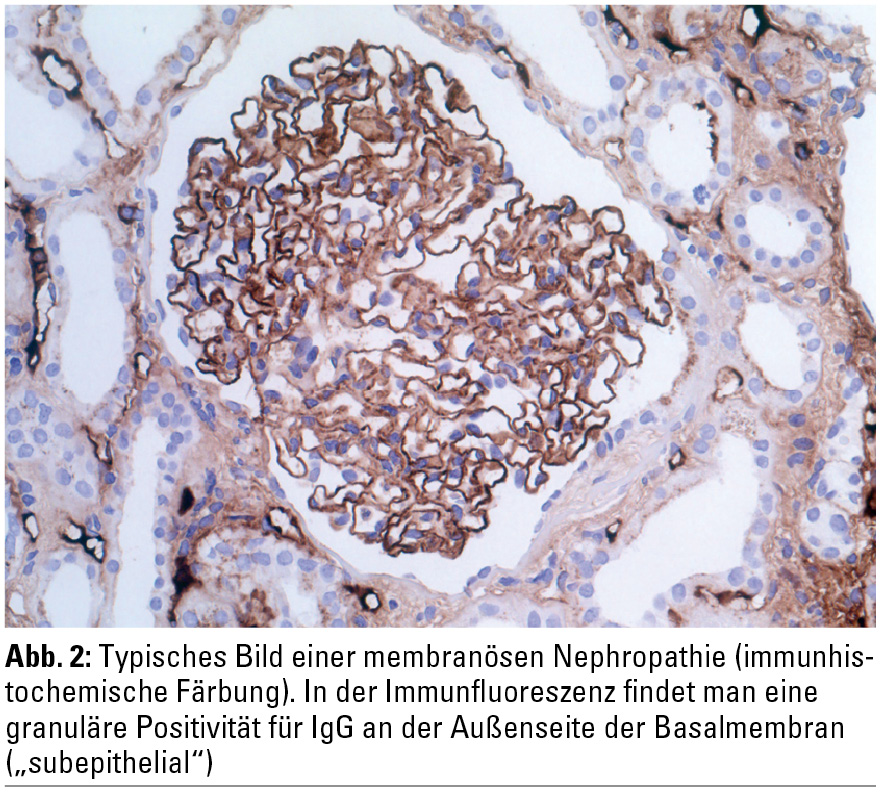
Die Diagnose einer MN wird histologisch gestellt. Deshalb sollte möglichst bei jedem Patienten mit einem nephrotischen Syndrom unklarer Genese eine Nierenbiopsie angestrebt werden. Beispiele für eine Silberfärbung und für eine routinemäßige lichtmikroskopische Färbung bei MN sind in den Abbildungen 1 und 2 (zur Verfügung gestellt von Dr. Dorothea Heininger) dargestellt. Zusätzlich zeigen sich in der Immunfluoreszenz für IgG und C3 positive granuläre Muster sowie in der Elektronenmikroskopie subepitheliale elektronendichte Ablagerungen und eine Expansion der glomerulären Basalmembran. Häufige sekundäre Ursachen für eine MN sind in der Tabelle zusammengefasst und sollten ausgeschlossen bzw. nachgewiesen werden. Antinukleäre Antikörper (ANA) und insbesondere Anti-dsDNA-Antikörper sind bei einer MN hinweisend für einen systemischen Lupus erythematodes (SLE) und eine Lupusnephritis der WHO-Klasse V. Eine chronisch-aktive Hepatitis B und seltener eine Hepatitis-C-Infektion wurden ebenso als eine sekundäre Ursache einer MN identifiziert. Die Patienten weisen serologisch meist eine Positivität für HBs-Antigen, HBc-Antikörper und HBe-Antigen auf. Es scheinen die HBe-Antigen-Antikörper-Immunkomplexe glomerulär abgelagert zu werden. Serumspiegel von Komplement C3 sind für gewöhnlich bei der MN im Normbereich, sie können aber insbesondere bei Lupus- und Hepatitis-B-assoziierter MN erniedrigt sein. Die Assoziation zwischen Malignomen und MN ist hinlänglich bekannt. Es wird angenommen, dass es zu einer subepithelialen Ablagerung des Tumorantigens mit nachfolgender Immunkomplexbildung, lokaler Komplementaktivierung und Podozytenschädigung kommt. Zu den häufigsten mit MN assoziierten Tumoren zählen solide (Prostata, Lunge, Gastrointestinaltrakt) oder hämatologische Malignome. Insbesondere bei Patienten über 65 Jahren, aber nicht nur bei ihnen, sollte daher bei Diagnose einer MN eine Tumorsuche gemäß den Richtlinien bzw. der klinischen Symptomatik durchgeführt werden (z. B. Labordiagnostik, Gastroskopie und Koloskopie, Mammographie bzw. Mammasonographie bei Frauen > 40 Jahren, PSA und eventuell urologische Untersuchung bei Männern > 50 Jahren, Thoraxröntgen und eventuell Thorax-CT etc.).
Eine genaue Medikamentenanamnese ist indiziert, da eine Assoziation zwischen der Einnahme bestimmter Medikamente und dem Auftreten einer MN beschrieben wurde. Zu diesen Therapeutika zählen u. a. Goldpräparate, Penicillamin, nichtsteroidale Antirheumatika, Anti-TNF-Therapien oder Captopril. Der pathogenetische Mechanismus dieser Assoziation ist nicht geklärt, aber es wird vermutet, dass eine T-Zell-abhängige polyklonale B-Zell-Aktivierung eine Rolle spielt.
Obwohl die exakte Bedeutung der PLA2R-Antikörper in der Pathogenese der IMN noch nicht geklärt ist, ist ein positiver serologischer Befund hoch suggestiv für das Vorliegen einer IMN. Gesunde Kontrollen und Patienten mit anderen nephrotischen Erkrankungen zeigen keine messbaren PLA2R-Antikörperspiegel. In Einzelfällen können zwar die PLA2R-Antikörper auch bei Patienten mit sekundärer MN detektiert werden, jedoch kann in diesen Fällen das zufällige Vorliegen einer IMN und einer anderen Erkrankung (SLE, Malignome, Hepatitis B) nicht gänzlich ausgeschlossen werden. Die derzeitige Datenlage erlaubt folgende Schlussfolgerungen:
- Die Höhe des PLA2R-Antikörperspiegels korreliert mit der Krankheitsaktivität.
- Bei einer Remission verschwindet auch der PLA2R-Antikörper.
- Ein Wiederauftreten könnte auf ein klinisches Rezidiv hindeuten.
In einer rezenten Studie konnten Hoxha und Kollegen zeigen, dass der lichtmikroskopische Nachweis des PLA2R-Antigens in den Glomerula mit einer idiopathischen, aber nicht mit einer sekundären Form einer MN assoziiert ist. Sowohl die PLA2R-Antikörperbestimmung im Serum als auch der Nachweis des PLA2R-Antigens in der Nierenbiopsie finden aktuell ihren Weg in die klinische Routine. Zurzeit werden die Spezifität und die Sensitivität dieser Tests und auch der Titerverlauf unter verschiedenen immunsuppressiven Therapien im klinischen Alltag von verschiedenen Forschungsgruppen untersucht.

Therapie und Verlauf
Im Juni 2012 wurden die KDIGO-Richtlinien für die Abklärung und Therapie der Glomerulonephritiden publiziert. Im Kapitel 7 wird die Abklärung und Therapie einer IMN erläutert. Da es bekanntermaßen in ca. einem Drittel aller Fälle von IMN zu einer Spontanremission kommen kann und diese für gewöhnlich in den ersten 6–12 Monaten nach der Diagnose auftritt, wird empfohlen, die Patienten mindestens 6 Monate lang optimal antiproteinurisch und antihypertensiv zu behandeln, bevor eine immunsuppressive Therapie begonnen wird. Eine Therapie mit ACE-Hemmern oder Angiotensin-Rezeptor-Blockern wird – ähnlich wie bei Patienten mit anderen proteinurischen Nierenerkrankungen – bei praktisch allen Patienten mit einer IMN empfohlen. Das Blutdruckziel ist < 130/80 mmHg oder sogar darunter. Patienten mit einem nephrotischen Syndrom aufgrund einer IMN und mit Serumalbumin < 2,5 g/dl haben ein signifikant erhöhtes Risiko für ein thromboembolisches Ereignis und sollten dementsprechend oral antikoaguliert werden.
Patienten sollten immunsuppressiv behandelt werden, wenn eines der folgenden Kriterien erfüllt ist:
- Proteinurie ist anhaltend > 4 g/Tag und 50 % über dem Ausgangswert und zeigt keine sinkende Tendenz trotz einer mindestens 6 Monate dauernden antihypertensiven und antiproteinurischen Therapie.
- Es treten schwere oder lebensgefährliche Komplikationen des nephrotischen Syndroms auf.
- Das Serumkreatinin steigt um > 30 % vom Ausgangswert innerhalb von 6-12 Monaten an und dieser Anstieg ist nicht durch Komplikationen erklärbar.
Es sollte keine immunsuppressive Therapie begonnen werden, wenn von einer chronischen Nierenfunktionseinschränkung auszugehen ist (Serumkreatinin > 3,5 mg/dl, eGFR < 30 ml/min/1,73m2, Nierengröße < 8 cm) oder bei Vorliegen von schweren oder lebensbedrohlichen Infektionen. Das Ziel einer immunsuppressiven Therapie ist die partielle Remission (PR, Proteinurie < 3,5 bis 4 g/Tag und 50%ige Reduktion vom Ausgangswert) oder die komplette Remission (CR, Proteinurie < 0,3 g/Tag), da gezeigt werden konnte, dass diese Patienten ein sehr gutes renales Langzeitüberleben haben.
Als initiale immunsuppressive Therapie wird Zyklophosphamid und Prednisolon nach dem Ponticelli-Schema empfohlen: 0,5 mg/kg Prednisolon täglich in den Monaten 1, 3 und 5 sowie Zyklophosphamid oral 2,0–2,5 mg/kg täglich in den Monaten 2, 4 und 6. Die Monate 1, 3 und 5 beginnen jeweils mit 1 g Methylprednisolon i. v. für 3 Tage ohne orales Prednisolon.
Unter dieser Therapie kommt es bei 72–88 % der Patienten zu einer CR oder PR. Der hohe Evidenzgrad (1B) dieser Empfehlung basiert auf drei positiven prospektiven, randomisierten und kontrollierten Studien zu diesem Schema. Der Erfolg einer solchen Therapie kann erst nach 6 Monaten beurteilt werden. Alternativ ist eine Therapie mit Zyklosporin A(3,5–5,0 mg/kg/Tag aufgeteilt auf 2 Tagesdosen) plus Prednisolon (0,15 mg/kg/Tag) oder Tacrolimus-Monotherapie (0,05–0,075 mg/kg/Tag aufgeteilt auf 2 Tagesdosen) möglich. Unter diesen Therapien wurden CR-/PR-Raten von 75–94 % beschrieben. Eine Therapie mit einem Kalzineurininhibitor (CNI) sollte für mindestens 6 Monate durchgeführt werden, wobei die optimale Therapiedauer unklar ist. Daten aus prospektiven Studien lassen bei responsiven Patienten eine Therapiedauer von mindestens 12 Monaten sinnvoll erscheinen. Bei gutem Ansprechen (CR oder PR) sollten die CNI-Dosen über einen Zeitraum von ca. 6 Monaten langsam ausgeschlichen werden. Kommt es jedoch innerhalb der ersten 6 Monate zu keinem Ansprechen auf eine CNI-Therapie, so sollte diese abgesetzt werden. Zyklophosphamid-Steroid- und Zyklosporin-Steroid-Kombinationstherapien sind vermutlich gleich effektiv, obwohl das Risiko eines Rezidivs des nephrotischen Syndroms unter einer CNI-Therapie höher ist (bis zu 60 % der Patienten nach Absetzen des CNI). Die Wahl des Therapieschemas hängt von verschiedenen Faktoren ab, z. B. der klinischen Erfahrung des Zentrums oder der Präferenz des Patienten. So wurde bisher an der Nephrologie Innsbruck hauptsächlich eine Therapie mit Zyklosporin A und Steroiden als initiale Therapie verwendet.
Eine Monotherapie mit Steroiden oder mit MMF sollte bei der IMN nicht durchgeführt werden. Bei Resistenz gegen eine Therapie mit Zyklophosphamid/Steroid empfehlen die KDIGO-Richtlinien einen Therapieversuch mit CNI ± Steroid und vice versa.
Im Licht der neuen pathogenetischen Erkenntnisse zum PLA2R-Antigen und zum PLA2R-Antikörper stellt Rituximab natürlich eine interessante Therapieoption dar. In den vergangenen Jahren wurden zunehmend Daten zu Rituximab entweder als Erstlinientherapie oder als Therapie bei resistenten Patienten publiziert. Die Ergebnisse von drei Beobachtungsstudien ohne Kontrollgruppe nach der Therapie mit Rituximab können wie folgt zusammengefasst werden:
- Die Wahrscheinlichkeit einer kompletten oder partiellen Remission liegt bei 53 bis 89 % und ist somit vergleichbar mit einer Zyklophosphamid- oder CNI-Therapie.
- Die Rituximab-Schemata 4-mal 375 mg/m2 in wöchentlichen Abständen oder 2-mal 1 g in 2-wöchigem Abstand sind vermutlich gleich effektiv. Im Zentrum Bergamo erfolgt generell eine einmalige Gabe von Rituximab 375 mg/m2. Die weitere Therapie wird anhand der B-Zell-Zahl im peripheren Blut gesteuert (Prof. Remuzzi, personal communication).
- Die Nebenwirkungen einer Rituximabtherapie sind vergleichsweise gering.
Obwohl noch keine prospektiven und randomisierten Studien zu Rituximab bei IMN vorliegen (eine Studie der Mayo Clinic zu diesem Thema wird erst 2016 fertig gestellt und ausgewertet sein; siehe auch www.clinicaltrials.gov), sind die vorhanden Daten sehr vielversprechend. Rituximab kann bei Patienten mit IMN, die auf eine initiale Standardtherapie nicht ansprechen oder schwere Nebenwirkungen erleiden, als alternative Therapie angesehen werden. Nichtsdestotrotz ist es derzeit komplett unklar, welches Rituximab-Therapieregime am effektivsten ist, ob eine begleitende immunsuppressive Therapie die Effektivität erhöhen könnte, ob die Rituximabtherapie in regelmäßigen Abständen oder aufgrund bestimmter Parameter (PLA2R-Antikörper-Flare?) wiederholt werden sollte und vor allem auch, welche Langzeitrisiken von einer Rituximabanwendung zu erwarten sind. Ein Abfall der PLA2R-Antikörperspiegel scheint einem klinischen Ansprechen auf eine immunsuppressive Therapie vorauszugehen und ebenso auch einen Relaps der Erkrankung (auch nach Transplantation) anzuzeigen. Aus diesem Grund scheint es sinnvoll, neben den üblichen Parametern Proteinurie und Nierenfunktion auch diesen Parameter zum Therapiemonitoring bei Patienten mit IMN zu verwenden.
Literatur beim Verfasser

AutorIn: OA Dr. Michael Rudnicki, MD FASN
Universitätsklinik für Innere Medizin IV – Nephrologie und Hypertensiologie, Medizinische Universität Innsbruck

Ursprünglich erschienen:
Neph 04|2012
Neph 04|2012