


























































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Rekurrenz der fokal segmentalen Glomerulosklerose nach Nierentransplantation
31. Dezember 2012
Die fokal segmentale Glomerulosklerose (FSGS), die in ihren histologischen Besonderheiten erstmals vor über 50 Jahren von Arnold Rich1 beschrieben wurde, zählt zu den häufigsten bioptischen Diagnosen beim nephrotischen Syndrom sowohl im Kindes- als auch im Erwachsenenalter.
Morphologisch präsentiert sich die FSGS zu Beginn mit strukturellen Veränderungen der Podozyten, mit Abflachung und Verlust der Fußfortsätze („effacement“) sowie deren Ablösung von der glomerulären Basalmembran („detachment“). Später kommt es zum charakteristischen Bild einer fokal und segmental verteilten glomerulären Sklerose. Etwa die Hälfte der betroffenen Patienten entwickelt eine progrediente Nierenfunktionsstörung, die im Schnitt innerhalb von 10–20 Jahren in eine terminale Niereninsuffizienz mündet. Neben einem kleineren Anteil sekundärer oder auch genetischer bzw. familiärer Formen lassen sich etwa 80 % der diagnostizierten Fälle als idiopathisch (primär) klassifizieren2, 3.
Größe und Persistenz einer Proteinurie sind starke Risikofaktoren für ein ungünstiges renales Outcome. So beschrieb Korbet4 in einer umfassenden Literaturübersicht, dass eine Proteinurie von über 10 g/24 h in der Mehrzahl der Fälle nach 3 Jahren mit einem terminalen Nierenversagen einhergeht.
Die Pathogenese der primären FSGS ist in ihrer Komplexität bislang nicht vollständig geklärt. Das heterogene Erscheinungsbild reflektiert eine Vielfalt verschiedener potenzieller Pathomechanismen, die der zentralen Pathologie einer Podozytenschädigung zugrunde liegen. Bei sporadischer primärer FSGS dürften zirkulierende Permeabilitätsfaktoren eine Rolle spielen. Als potenzielle Kandidaten werden nun der soluble Urokinase-Typ Plasminogenaktivator-Rezeptor (suPAR) sowie die Zytokine Tumornekrosefaktor-alpha (TNF-α) bzw. Cardiotrophin-like Cytokine-1 (CLC-1) diskutiert5–7.
Andererseits sind familiäre Formen (autosomal rezessiver bzw. dominanter Erbgang) und auch Formen mit Mutationen in verschiedenen Genen beschrieben, die eine Beeinträchtigung der podozytären Struktur und Funktion bzw. der Integrität der glomerulären Filtrationsbarriere bedingen. FSGS-assoziierte Mutationen können eine Vielzahl unterschiedlicher Gene betreffen, unter anderem die Gene NPHS1 oder NPHS2, deren Proteine Nephrin und Podocin in das Split-Diaphragma eingelagert sind. Die rezente Literatur zeigt die besondere Komplexität der FSGS-Genetik auf. Kürzlich wurde z. B. für Afroamerikaner auch eine Assoziation mit ApoL1-Varianten gezeigt8. Auch wurde aktuell eine Assoziation einer FSGS-Variante mit Polymorphismen in Faktoren des alternativen Komplementwegs, wie Faktor H und C3 berichtet9.
Die sekundäre FSGS hat mannigfaltige Ursachen. Sie kann virusassoziiert (z. B. HIV), drogen- und medikamenteninduziert oder adaptiv infolge glomerulärer Hyperfiltration und erhöhtem glomeruläre Druck auftreten2.
In unterschiedlichen Ätiologien begründet sich auch die nicht einheitliche Relevanz verschiedener Formen für den Verlauf nach Nierentransplantation.
Wann und wie oft kommt es zu einer FSGS-Rekurrenz?
Bei 30–50 % der Patienten, die aufgrund einer durch FSGS bedingten terminalen Niereninsuffizienz transplantiert werden, ist die FSGS-Rekurrenz die primäre Ursache eines Transplantatverlusts. Eine Rekurrenz, welche sich oft durch Proteinurie, Hypertension und/oder eine progrediente Nierenfunktionsverschlechterung manifestiert, tritt bei Kindern im Durchschnitt schon zwei Wochen und bei Erwachsenen Monate nach Transplantation auf10, 11. In einer rezenten Studie konnte gezeigt werden, dass im Vorfeld einer Rekurrenz bereits unmittelbar nach Transplantation in Reperfusionsbiopsien ein „effacement“ beobachtet werden kann12. Lichtmikroskopisch kann sich eine Rekurrenz in sehr frühen Stadien noch unauffällig präsentieren. Das Abwarten einer bioptisch gesicherten Diagnose kann den Therapiebeginn beträchtlich verzögern. Die Biopsie ist für die definitive Diagnose einer FSGS-Rekurrenz und für den sicheren Ausschluss anderer Differenzialdiagnosen jedoch unerlässlich13.
Wer hat das Risiko?
Risikofaktor für eine FSGS-Rekurrenz ist ein früher Krankheitsbeginn in der Kindheit. Bei 50–80 % der unter 6-Jährigen kommt es zur Rekurrenz, einem schnellen Voranschreiten hin zum terminalen Nierenversagen oder Formen mit diffuser mesangialer Zellvermehrung (Hyperzellularität)14. Nach Organverlust aufgrund einer rekurrenten FSGS ist das Rekurrenzrisiko besonders hoch. Die Wahrscheinlichkeit einer neuerlichen Rekurrenz in einem weiteren Transplantat liegt nahezu bei 100 %15. Zudem wurde in einer großen retrospektiven pädiatrischen Studie mit 6.484 Kindern ein signifikant erhöhtes Rekurrenzrisiko für Empfänger einer Lebendspende beschrieben16.
Was gibt es Neues zur Pathogenese?
Das rasche Auftreten rekurrierender FSGS nach Transplantation sowie das in vielen Fällen gute Ansprechen auf Plasmapherese begründet die Vermutung einer pathogenetischen Rolle zirkulierender Permeabilitätsfaktoren. Bereits 1991 berichteten Lagrue et al.17 über die spontane Remission einer Proteinurie bei neugeborenen Kindern einer an FSGS erkrankten Mutter und stellten die Hypothese eines zirkulierenden humoralen Faktors auf. Kürzlich beschrieben Gallon et al.18 den Fall einer schweren refraktären FSGS-Rekurrenz, wobei hier die transplantierte Niere ektomiert und das Organ in einen Patienten mit diabetischer Nephropathie weiter transplantiert wurde. Nach dieser neuerlichen Transplantation kam es zu einer prompten Stagnation der Proteinurie und zur Rückbildung FSGS-typischer morphologischer Läsionen.
Frühe Studien zeigten eine mögliche Bedeutung eines ≤ 50 kDa-Faktors im Serum erkrankter Patienten, der als Trigger der für die FSGS typischen Schädigungen des glomerulären Filters und Erhöhung der Membranpermeabilität für Albumin wirken könnte19.
Wei et al.5 konnten nun in einer hochkarätig publizierten Studie anhand diverser experimenteller und klinischer Daten eine pathogenetische Rolle des zirkulierenden (solublen) Urokinaserezeptors suPAR zeigen. Die Autoren beschreiben, dass dieser Rezeptor in seiner löslichen Form direkte ungünstige Effekte auf Podozyten entfalten kann, offenbar über eine Aktivierung des für die Anhaftung von Podozyten an die Basalmembran so essenziellen Adhäsionsmoleküls b3-Integrin. Dieser Effekt bedingt, wie auch in einigen komplexen Mausmodellen beschrieben werden konnte, eine Störung der glomerulären Filtrationsbarriere mit dem Resultat einer Proteinurie. Eindrucksvoll war die häufige und für das Krankheitsbild hochspezifische Erhöhung von suPAR im untersuchten Patientenkollektiv (70 % der 63 untersuchten Patienten mit primärer FSGS), wobei erhöhte Spiegel mit einer Rekurrenz nach Transplantation assoziiert waren. Ein Ansprechen auf Plasmapherese war dabei mit dem Rückgang von suPAR-Blutkonzentrationen und der Aktivität von β3-Integrin assoziiert5. Die Beziehung zwischen suPAR und dem von Savin et al.19 erstbeschriebenen Permeabilitätsfaktor bleibt allerdings derzeit noch unklar.
Weiters gibt es klinische und experimentelle Hinweise für eine mögliche Rolle von TNF-α als FSGS-Trigger. Leroy et al.20 berichteten zum Beispiel den Fall eines Kindes, welches nach Anti-TNF-α-Therapie eine Remission seiner rekurrierenden FSGS erreichte. Die Ergebnisse experimenteller Studien lassen vermuten, dass TNF-α seine Effekte auch über eine Aktivierung von β3-Integrin triggern könnte. Es stellt sich die Frage nach funktionellen Zusammenhängen zwischen TNF-α, suPAR und b3-Integrin6.
Bei familiärer FSGS sollte das Risiko einer Rekurrenz nicht gegeben sein, zumindest bei normalem genetischem Background des Spenders. In diesem Zusammenhang sind rezente Daten zu NPHS2, einem Gen, das für das integrale podozytäre Membranprotein Podocin kodiert, bedeutsam. NPHS2-Mutationen wurden erstmals bei der familiären autosomal-rezessiven FSGS und in sporadischen Fällen des steroidresistenten nephrotischen Syndroms beschrieben21. In einer Kohorte von 83 Patienten konnten nun Jungraithmayr und Mitarbeiter22 zeigen, dass Patienten mit einer homozygoten oder compound-heterozygoten Mutation im NPHS2-Gen nicht zu einer FSGS-Rekurrenz nach Transplantation neigen. Demzufolge könnte eine genetische Testung im Vorfeld einer geplanten Transplantation helfen, das Risiko einer Rekurrenz abzuschätzen. Nichtsdestotrotz sind, wenn auch viel seltener, Rekurrenzen für genetische Varianten beschrieben. Zudem bleibt zu erwähnen, dass ein beträchtlicher Anteil der Patienten mit einer NPHS1-Mutation eine De-novo-Glomerulonephritis entwickelt. Der Mechanismus ist hier ein Podozytenschaden durch Anti-Nephrin-Antikörper, die gegen das transplantierte Organ gerichtet sind23.
Welche Therapieoptionen gibt es?
Die Therapie rekurrenter FSGS ist eine besondere Herausforderung, denn große randomisiert kontrollierte Studien, in denen verfügbare Konzepte systematisch untersucht wurden, sind nicht verfügbar. Im Wesentlichen beruht unser derzeitiges Wissen auf Ergebnissen aus Fallberichten und Fallserien. Publizierte Therapieschemen sind dabei sehr heterogen, und aufgrund einer gewissen Polypragmasie ist die Effizienz individueller Maßnahmen oft schwierig festzumachen.
Ein wesentliches Standbein in der Therapie der rekurrenten FSGS ist die Apheresetherapie, hier vor allem die Plasmapherese. Primärer Wirkmechanismus ist wahrscheinlich die Elimination postulierter zirkulierender Faktoren, wie in der bereits erwähnten Arbeit von Wei et al.5 auch für suPAR gezeigt werden konnte. In Kombination mit anderen therapeutischen Prinzipien, wie z. B. Zyklophosphamid, hoch dosiertem CyA, Steroidtherapie und/oder Rituximab wurde Plasmapherese bei rekurrenter FSGS als Prophylaxe oder Therapie – auch bei relapsierenden Verläufen – erfolgreich eingesetzt (partielle und komplette Remission bei etwa 40–100 % der behandelten Patienten). Dauer und Frequenz einer solchen Therapie waren dabei sehr unterschiedlich und reichten von 5 bis über 50 Behandlungssitzungen. Insbesondere in frühen Stadien einer Rekurrenz scheint Plasmapherese besonders effektiv zu sein13.
Unkontrollierte Daten weisen darauf hin, dass bestimmte Anpassungen der Basisimmunsuppression, parallel zur Plasmapherese, effizient sein könnten. Ein möglicher Ansatz ist z. B. eine hoch dosierte orale oder intravenöse Zyklosporin-A-Therapie (Talspiegel: 200–400 ng/ml; C2-Spiegel: 1.200–1.400 ng/ml)24. Der antiproteinurische Effekt von Zyklosporin A scheint dabei über direkte Effekte am Glomerulum vermittelt zu sein25.
Als weitere Option wurde (in Prophylaxe und Therapie) Rituximab eingesetzt. Der Wirkmechanismus von Rituximab als chimärer murin-humaner Anti-CD20-Antikörper bleibt im Kontext der rekurrierenden FSGS vorerst unklar, und Mechanismen, wie z. B. Blockade einer B-Zell-vermittelten Freisetzung von Permeabilitätsfaktoren, bleiben spekulativ26. Rituximab könnte unabhängig von CD20, über Bindung der SMPDL-3b (Sphingomyelin Phosphodiesterase, acid-like 3b) und Regulation der Acid-Sphingomyelinase(ASMase)-Aktivität, einen günstigen Effekt auf das Aktin-Zytoskelett der Podozyten haben. Fornoni et al.27 konnten bei 27 Patienten mit hohem Risiko für eine FSGS-Rekurrenz zeigen, dass Rituximab einer (vermutlich durch einen Permeabilitätsfaktor vermittelten) Reduktion von SMPDL-3b und ASMase entgegenwirken und somit das Auftreten einer Proteinurie verhindern kann. Eine Prävention von FSGS-Rekurrenz konnte auch von anderen Autoren für Einzelfälle und Fallserien berichtet werden (Tab. 1). Unkontrollierte klinische Erfahrungsberichte zeigen zudem, dass einige, wenn auch nicht alle Patienten mit manifester rekurrenter FSGS auf eine Rituximab-Therapie mit zumindest partieller Remission ansprechen (Tab. 2).
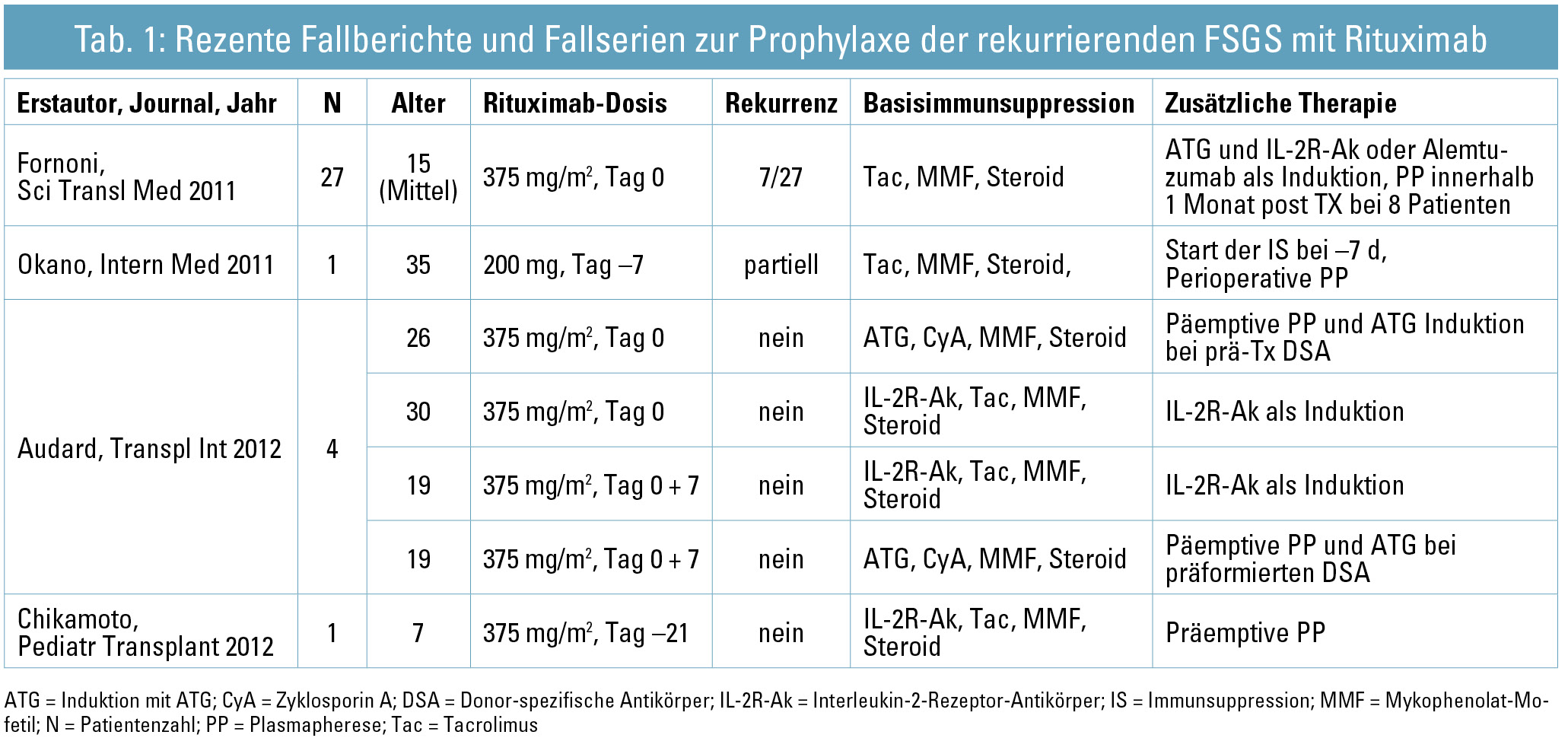
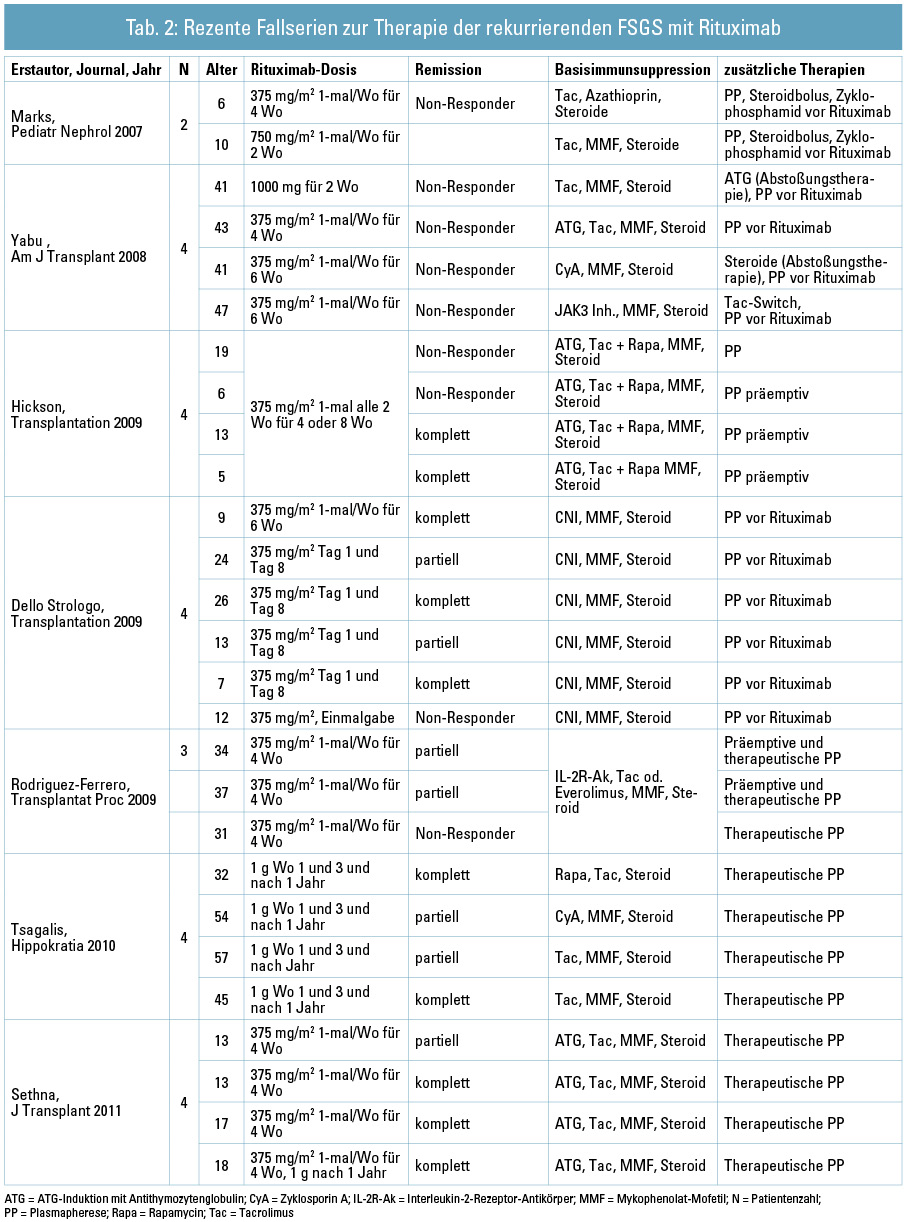
Ein vielversprechender therapeutischer Ansatz ist zudem, wie schon oben erwähnt, der Einsatz von TNF-α-Antagonisten. In einem aktuellen Fallbericht konnten zum Beispiel Bitzan et al.28 bei einem Patienten mit Plasmapherese-refraktärer FSGS-Rekurrenz unter wiederholten Gaben von Etanercept (später Switch auf Infliximab) eine partielle Remission mit Reduktion der Eiweißausscheidung von 10 auf unter 2 g/24 h erreichen.
Wenn auch nicht für die Sondersituation der rekurrenten FSGS angelegt, möchten wir abschließend zwei aktuelle prospektive Studien (Rekrutierungsphase) erwähnen, deren Ergebnisse auch für die Rekurrenz interessant sein könnten. So läuft eine große multizentrische Phase-II-Studie (FONT), die die Wirkung von Adalimumab, einem monoklonalen Antikörper gegen TNF-α, versus oraler Galaktose (postulierte Bindung und Neutralisation des zirkulierenden Permeabilitätsfaktors) versus konservativer Therapie allein (Lisinopril, Losartan, Atorvastatin) in der Behandlung der therapieresistenten FSGS (NCT00814255) untersucht. Kürzlich wurde zudem eine weitere Studie zum Effekt oraler Galaktose auf den Verlauf einer FSGS registriert (NCT01113385). Es bleibt zu hoffen, dass diese und weitere Studien, in denen neue Therapiekonzepte systematisch geprüft werden, eine solide Basis für einen effizienten und evidenzbasierten Einsatz therapeutischer Strategien ermöglichen.
1 Bull John Hopkins Hosp 1957; 100:173
2 D’Agati et al., N Engl J Med 2011; 365:2398
3 Shimizu et al., Clin Transplant 2011; 25 Suppl 23:6
4 Korbet, Nephrol Dial Transpl 1999; 14 Suppl 3:68
5 Wei et al., Nature Med 2011; 17:952
6 Bitzan et al., Pediatr Nephrol (in press)
7 McCarthy et al., Clin Am J Nephrol 2010; 5:2115
8 Genovese et al., Science 2010; 329:841
9 Sethi S et al., Am J Kidney Dis 2012; 60:316
10 Artero et al., Am J Med 1992; 92:375
11 Chadban et al., J Am Soc Nephrol 2001; 12:394
12 Chang et al., Transplantation (in press)
13 Pradhan et al., Pediatr Nephrol 2003; 18:934
14 Shimizu et al., Clinic Transplant 2011; 25 Suppl 23:6
15 Jungraithmayr et al., J Am Soc Nephrol 2011; 22:57
16 Baum et al., Kidney Int 2001; 59:328
17 Lagrue et al., Presse Med 1991; 20:255
18 Gallon et al., New Engl J Med 2012; 366:1648
19 Savin et al., New Engl J Med 1996; 334:878
20 Leroy et al., Am J Transplant 2009; 9:858
21 Boute et al., Nat Genet 2000; 24:349
22 Jungraithmayr et al., J Am Soc Nephrol 2011; 22:579
23 Vinai et al., Pediatr Transplant 2010; 14:314
24 Ulinski et al., Curr Opin Organ Transplant 2010; 15:628
25 Faul et al., Nat Med 2008; 14:931
26 Haffner et al., Pediatr Nephrol 2009; 24:1433
27 Fornoni et al., Sci Transl Med 2011; 3:85ra46
28 Bitzan et al., Pediatr Nephrol (in press)

AutorIn: Dr. Elisabeth Schwaiger
Abteilung für Nephrologie und Dialyse, Universitätsklinik für Innere Medizin III, Wien

AutorIn: Univ.-Prof. Dr. Georg A. Böhmig
Abteilung für Nephrologie und Dialyse, Universitätsklinik für Innere Medizin III, Wien

Ursprünglich erschienen:
Neph 04|2012
Neph 04|2012