
























































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Niere und RAS* – Kontroverses, Provokantes und Neues
9. Dezember 2011
Kontroverses und Provokantes
Die wichtigsten derzeit gültigen Richtlinien empfehlen bei Patienten mit chronischer Niereninsuffizienz eine oft drastische Blutdrucksenkung in den normotensiven Bereich. RAS(Renin-Angiotensin- System)-Blocker gelten als First-Line-Therapie, weil ihnen zusätzlich zur blutdrucksenkenden Wirkung ein günstiger Effekt auf die Progression der Niereninsuffizienz zugeschrieben wird („Nephroprotektion“). Dies trifft insbesondere für Patienten mit großer Proteinurie zu.
Wie nephroprotektiv sind RAS-Hemmer?
Nachdem es über knapp zwei Dekaden fast als Kunstfehler gegolten hat, einem Nierenpatienten keinen RAS-Blocker zu verschreiben, scheint nun eine gewisse Ernüchterung hinsichtlich der nephroprotektiven Wirkung der RAS-Hemmer einzutreten. Die vorliegende Arbeit soll auch als Anstoß dienen, sich kritisch mit etablierten Meinungen auseinanderzusetzen und manche Dogmen zu hinterfragen.
Vor wenigen Wochen erschien in der Cochrane Database ein Review über „ACE-Hemmer und Angiotensinrezeptorblocker (ARB) bei Erwachsenen mit früher (KDOQI Stadium 1–3), nicht diabetischer chronischer Niereninsuffizienz“. Die Autoren identifizierten lediglich vier randomisierte und kontrollierte Studien, die ihren Einschlusskriterien entsprachen. Die vorliegenden Daten belegen ihrer Meinung nach weder einen günstigen Einfluss von ACE-Hemmern oder ARB auf die Gesamtmortalität noch auf kardiovaskuläre Ereignisse noch auf das Risiko, eine terminale Niereninsuffizienz zu entwickeln. Bei den Patienten im Stadium 1–3 handelt es sich, wie wir aus vielen epidemiologischen Studien wissen, um die weitaus größte Gruppe der chronisch Nierenkranken!
„Überhaupt hat der Fortschritt das an sich, dass er viel größer ausschaut, als er wirklich ist.“
Johann Nepomuk Nestroy
Spezialist für Evidence-based Medicine, 1801–1862
Im Jahr 2005 wurde eine groß angelegte Metaanalyse veröffentlicht, die den Effekt von RASHemmern auf renale Endpunkte untersuchte. Placebokontrollierte Studien zeigten zwar einen deutlichen Vorteil der RAS-Hemmer, der allerdings durch den niedrigeren Blutdruck unter RAS-Hemmung erklärbar war. Beim direkten Vergleich zwischen RAS-Hemmern und anderen antihypertensiven Medikamenten ergab sich zwar eine relative Risikoreduktion von 29% (Relative Risk 0,71) für die Verdoppelung des Serumkreatinins, allerdings mit einem weiten 95%-Konfidenzintervall von 0,49–1,04. Das relative Risiko für den Endpunkt terminale Niereninsuffizienz (ESRD) wurde um 13 % verringert (RR 0,87, 95% CI 0,75– 0,99). Überraschenderweise fand diese Metaanalyse keinen Vorteil für RAS-Hemmer bei der diabetischen Nephropathie. Das relative Risiko für die Verdoppelung des Serumkreatinins lag hier bei 1,09 (95% CI 0,55–2,15), für das Erreichen der terminalen Niereninsuffizienz bei 0,89 (95% CI 0,74–1,07). Diese Studie von Casas wurde vom „scientific establishment“ massiv kritisiert, weil die Daten der großen ALLHAT-Studie (Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial) in die Analyse einflossen, die keinen primären renalen Endpunkt hatte. Allerdings ergab eine Subgruppenanalyse der diabetischen Population in der ALLHAT-Studie, dass mehr Patienten mit Lisinopril eine terminale Niereninsuffizienz entwickelten als in der Chlortalidon-Gruppe (25/1.563 vs. 26/2.755; p = 0,05, RR 1,74, 95% CI 1,00–3,01).
Mehr RAS-Hemmer, mehr Dialysepatienten
Zweifel an der nephroprotektiven Wirkung der RAS-Hemmer tauchten auch von anderen Seiten auf. In den Vereinigten Staaten wurde zwischen 1993 und 2000 ein dramatischer Anstieg der Dialysepatienten mit diabetischer Nephropathie verzeichnet, der zu weniger als 10% durch die steigende Prävalenz des Diabetes in der Bevölkerung oder ein verbessertes kardiovaskuläres Überleben erklärbar war. Der Anstieg wurde beobachtet, „obwohl“ in diesem Zeitraum bereits vermehrt nephroprotektive Medikamente (ACE-Hemmer) eingesetzt wurden. Bekanntlich war 1993 die Arbeit der Collaborative Study Group über den protektiven Effekt von Captopril auf die diabetische Nephropathie erschienen.
Eine kanadische Kohortenstudie zeigt, dass für Diabetiker das relative Risiko, die terminale Niereninsuffizienz zu erreichen, unter ACE-Hemmern im Vergleich zu Thiaziden auf das 2,5- Fache erhöht ist (95% CI 1,3–4,7). Für Betablocker lag das relative Risiko bei 0,8 (95% CI 0,5–1,4) und für Kalziumantagonisten bei 0,7 (95% CI 0,4–1,3). Bemerkenswerterweise lag das relative Risiko für ACE-Hemmer während der ersten drei Jahre bei 0,8 (95% CI 0,3–2,5) und stieg danach auf 4,2 (95% CI 2,0–9,0). Dies lässt den hypothetischen Schluss zu, dass ACE-Hemmer zeitlich begrenzt eine günstige (oder zumindest keine negative) Wirkung auf die Progression der Nierenerkrankung ausüben und möglicherweise langfristig mehr schaden als nutzen. Die Dauer der meisten RAS-Hemmer-Studien überschreitet den Zeitraum von drei Jahren nicht.
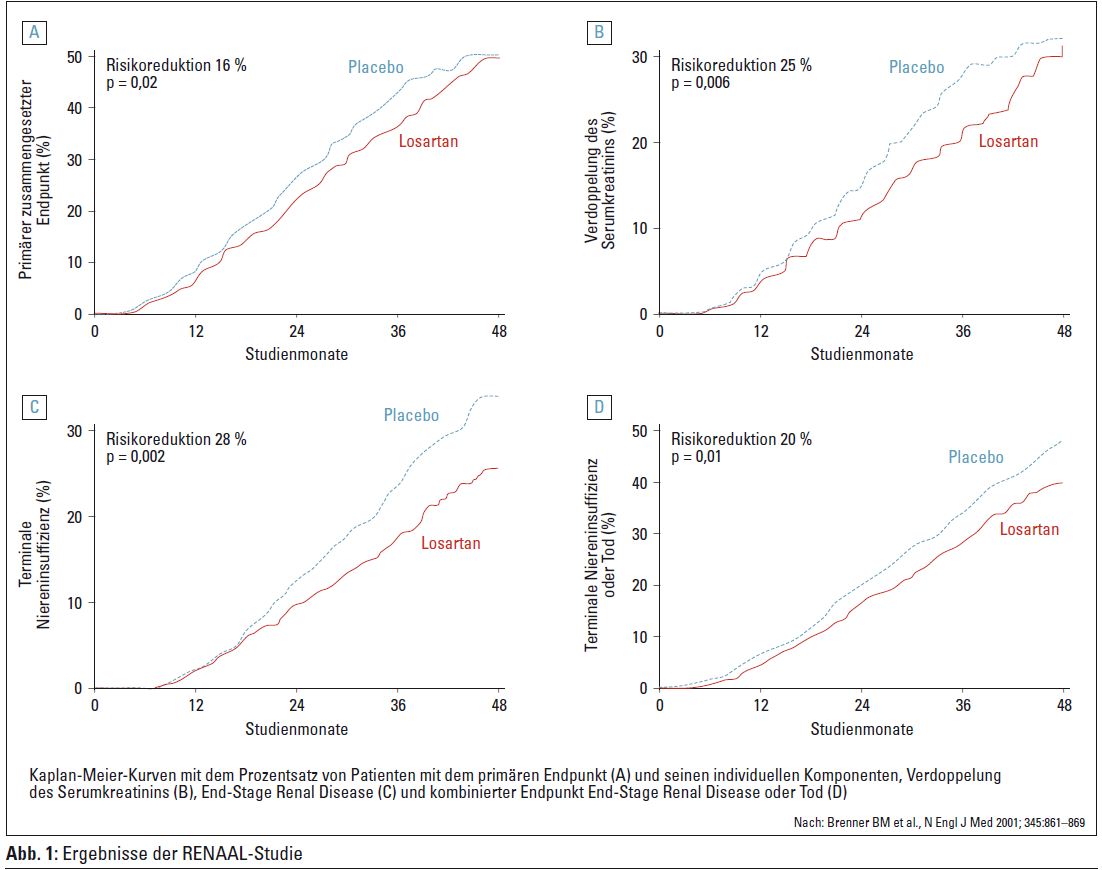
RENAAL kritisch betrachtet
In diesem Zusammenhang lohnt es sich auch, die RENAALStudie (Auswirkung von Losartan auf renale und kardiovaskuläre Endpunkte bei Typ-2-Diabetikern mit diabetischer Nephropathie) genauer anzusehen. In Abbildung 1 ist gut zu erkennen, dass sich nach vier Jahren die Kurven des primären Endpunkts (zusammengesetzt aus Verdoppelung des Serumkreatinins, Erreichen der terminalen Niereninsuffizienz und Tod) von Losartan und Placebo weitgehend annähern. Ähnlich verhält es sich bei der Verdoppelung des Serumkreatinins. Lediglich beim Erreichen der terminalen Niereninsuffizienz bleibt der Unterschied auch nach vier Jahren erhalten. Da in der Losartan-Gruppe numerisch (statistisch nicht signifikant) mehr Todesfälle als in der Placebogruppe auftraten, wählten die Autoren für die Abbildung nicht Tod als einen der sekundären Endpunkte, sondern kreierten einen neuen, zusammengesetzten Endpunkt „Tod und terminale Niereninsuffizienz“. Sonst wäre die Kurve für Losartan über der Placebokurve zu liegen gekommen. Ein gutes Beispiel dafür, wie man Daten manipulieren kann, ohne zu schwindeln.
Metaanalysen in Diskussion
In den vergangenen zehn Jahren wurden insgesamt drei Metaanalysen auf Patientenebene von elf randomisierten und kontrollierten Studien publiziert, die zeigten, dass auch nach Adjustierung der Daten in Bezug auf Blutdruck und Proteinurie ein Vorteil für die ACE-Hemmung zu verzeichnen ist (RR 0,67, 95% CI 0,53–0,84). Allerdings blieben diese Metaanalysen nicht unwidersprochen: Zwei der elf Studien sind nicht publizierte „persönliche Mitteilungen“, das mittlere Alter der Studienpopulation lag bei 52 Jahren (Bereich 46–63 Jahre) und entspricht somit nicht dem Hauptkollektiv, mit dem wir es zu tun haben, außerdem betrug die mittlere Studiendauer nur 2,2 Jahre (Bereich 0,9–2,4 Jahre). Methoden und Frequenz der Quantifizierung der Proteinurie differierten beträchtlich, ebenso wenig gab es ein standardisiertes Protokoll für die Blutdruckmessung.
Eine ähnlich kritische Zusammenfassung der Studienlage zur Nephroprotektion der RAS-Hemmer wurde 2006 von Griffin und Bidani publiziert. Die Hauptwirkung der RAS-Blocker beruht ihrer profunden Analyse nach in erster Linie auf der Blutdrucksenkung. Sie kommen zu dem Schluss, dass die der RAS-Blockade zugeschriebene renoprotektive Wirkung stark übertrieben dargestellt wird.
Neues zum RAAS
In den vergangenen 20 Jahren hat sich, vielfach unbemerkt vom klinischen Alltag, das Verständnis des Renin-Angiotensin- Aldosteron-Systems (RAAS) deutlich verändert. Wir stehen gleichsam vor einem Paradigmenwechsel. Das traditionelle Bild des RAAS erwies sich zwar nicht als falsch, es ist jedoch durch neue Erkenntnisse um ein Vielfaches komplexer geworden.
Traditionelles Verständnis des RAAS Das von Tigerstedt und Bergman bereits 1898 entdeckte Renin wird als Prorenin in den juxtaglomerulären Zellen der afferenten Arteriole gebildet und in Sekretgranula im Golgi-Apparat gespeichert. Zum Teil erfolgt dort bereits die Umwandlung in Renin, es gibt allerdings auch extrarenale Orte, wo Prorenin zu Renin konvertiert werden kann. Während man bisher annahm, dass Prorenin keine eigene intrinsische Aktivität entfaltet, hat sich diese Ansicht mit der Entdeckung von Proreninrezeptoren (siehe später) geändert.
Zu den wichtigsten Stimuli für die Freisetzung von Renin aus dem juxtaglomerulären Apparat zählen eine glomeruläre Hypoperfusion, hervorgerufen durch Hypotonie oder Volumendepletion, eine niedrige Na-Konzentration im distalen Tubulus und eine vermehrte Aktivität des sympathischen Nervensystems.
Von dem in der Leber gebildeten Angiotensinogen, einem Alpha-2-Globulin bestehend aus 452 Aminosäuren, wird in einem ersten Schritt durch das proteolytische Enzym Renin ein aus zehn Aminosäuren bestehendes Peptid abgespalten – Angiotensin I. Von diesem Peptid werden durch das Angiotensin-Converting Enzyme (ACE, auch Kininase II genannt) nochmals zwei Aminosäuren entfernt, wodurch das Oktapeptid Angiotensin II entsteht. ACE befindet sich hauptsächlich in der Lunge, ist aber auch im Plasma und im Interstitium nachweisbar, außerdem findet sich ein Gewebs-ACE in allen wichtigen Organen.
Als einer der stärksten Vasokonstriktoren erhöht Angiotensin II den Blutdruck und stimuliert zusätzlich die Freisetzung von Aldosteron aus der Zona glomerulosa der Nebennieren. Aldosteron bewirkt eine vermehrte Na- und Wasserrückresorption am distalen Tubulus. Angiotensin II greift auch in die Regulation des sympathischen Nervensystems und in den Durstmechanismus ein. Die wichtigsten physiologischen Wirkungen von Angiotensin II werden über den AT1-Rezeptor mediiert: Vasokonstriktion, Hypertrophie und Hyperplasie der Gefäßzellen, Na-Retention, Bildung von Sauerstoffradikalen, Auslösung von inflammatorischen, thrombotischen und fibrotischen Prozessen. Über den AT2-Rezeptor werden unter physiologischen Bedingungen die wichtigsten AT1-Rezeptor-mediierten Wirkungen antagonisiert.
Gegenwärtiges Verständnis des RAAS
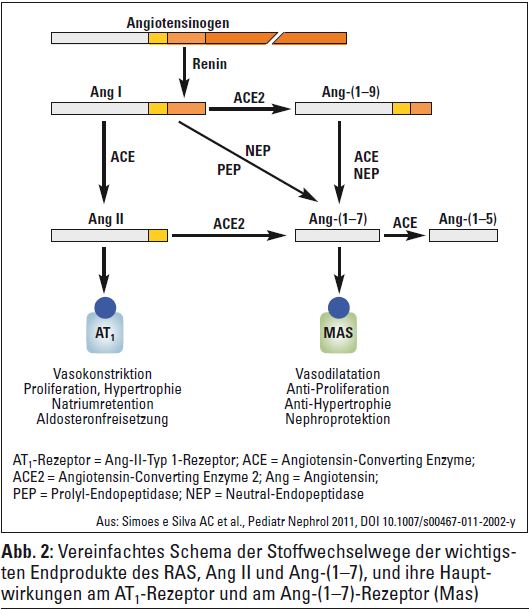
Neben dem lokal wirksamen RAAS (Gehirn, Herz, Blutgefäße, Niere, Pankreas usw.) wurden in den letzten Jahren auch neue, biologisch aktive Peptide und spezifische Rezeptoren entdeckt, deren klinische Relevanz derzeit noch nicht genau feststeht, die aber weitere Mosaiksteine auf dem Weg zum Verständnis dieses komplexen blutdruckregulierenden Systems darstellen. Prorenin galt bislang als inaktive Vorstufe von Renin, das als proteolytisches Schlüsselenzym des RAAS vom Vorläufer Angiotensinogen das Dekapeptid Angiotensin I abspaltet. 2002 publizierten Nguyen et al. die Entdeckung des Proreninrezeptors, der, wie sein Name bereits nahelegt, sowohl Prorenin als auch Renin bindet und über einen intrazellulären Signalweg zwei MAP-Kininasen (ERK 1 und 2) aktiviert. Unabhängig von Angiotensin II werden hierdurch profibrotische und COX2-Gene aktiviert, sodass über diesen Weg möglicherweise Organschäden bei Diabetes und Hypertonie erklärbar sind. ACE ist nach neueren Erkenntnissen nicht nur als hydrolytisches Enzym für die Bildung von Angiotensin II verantwortlich, sondern agiert nach Bindung eines ACE-Hemmers bzw. auch von Bradykinin als Auslöser einer intrazellulären Signalkaskade, die zu vermehrter Synthese von ACE und Cyclooxygenase- 2 führen kann. Auch hier sind der Stellenwert und die klinische Relevanz dieser Entdeckung noch nicht geklärt. Bemerkenswerterweise wurde im Jahr 2000 eine Captopril-insensitive Carboxypeptidase entdeckt, die später ACE2 genannt wurde. Ursprünglich wurde angenommen, dass dieses Enzym ebenfalls Angiotensin I – allerdings zu Angiotensin-(1–9) – hydrolysiert. Erst später fand man heraus, dass ACE2 hauptsächlich Angiotensin II als wichtigstes Substrat zu Ang-(1–7) hydrolysiert. Bei vielen Nierenerkrankungen wurde eine verminderte Expression von ACE2 in den Nieren festgestellt, wodurch es lokal zur Erhöhung der Angiotensin-II-Konzentration kommt. Diese wiederum könnte für die Entstehung von Nierenschäden mitverantwortlich sein. Sowohl ein erworbener wie auch ein genetischer Mangel an ACE2 führen zu Albuminurie und Nierenschädigung.
Rolle von Ang-(1–7)? Über viele Jahre hindurch wurde Ang- (1–7) als inaktives Peptid angesehen, das durch einen ACE unabhängigen Mechanismus aus Angiotensin I entsteht. Mit der Entdeckung von ACE2 wurde Angiotensin II als eigentliches Substrat dieses Enzyms identifiziert. Das dadurch entstehende Heptapeptid Ang-(1–7) wirkt über einen spezifischen Rezeptor (Mas-Rezeptor) gleichsam als Gegenspieler von Angiotensin II. Es hebt die vasokonstriktorische und proliferative Wirkung von Angiotensin II auf und wirkt antiarrhythmogen und antithrombogen. In der Niere gibt es sowohl glomeruläre als auch tubuläre Angriffspunkte des Ang-(1–7). Häufig, aber nicht immer, fungiert es dort als Gegenspieler von Angiotensin II. Neben einer vasodilatatorischen Wirkung an den afferenten Arteriolen (Erhöhung des renalen Blutflusses) hat es über den Mas-Rezeptor auch einen natriuretischen Effekt im proximalen Tubulus. Etwas verwirrend, kann es allerdings auch einen antidiuretischen Effekt (ebenfalls über den Mas-Rezeptor) aufweisen. In tierexperimentellen Untersuchungen wurde eine Reihe von weiteren zum Teil kontradiktorischen Effekten beschrieben.
Ang-(1–7) scheint auch eine wichtige Rolle in der Pathophysiologie der primären Hypertonie zu spielen. Auch im Endstadium der chronischen Niereninsuffizienz ist Ang-(1–7) erhöht. Allerdings ist es noch zu früh, die in Tierexperimenten und zum Teil auch beim Menschen gemachten Beobachtungen zu einem kohärenten Bild zusammenzufassen.
Literatur beim Verfasser
* Auf Aldosteron wird in dieser Arbeit bewusst nicht eingegangen, da im vorliegenden Heft Ass. Dr. Hildegard Hafner-Gießauf und Univ.-Prof. Dr. Alexander Rosenkranz dieses Thema behandeln.
NEPHRO Spot
Das für die Homöostase des Körpers so wichtige Renin-Angiotensin-Aldosteron(RAAS)-System besitzt zwei entgegengesetzte Komponenten: einerseits das durch Angiotensin-Converting Enzyme (ACE) entstehende Angiotensin II, das seine Hauptwirkungen über den AT1-Rezeptor ausübt, andererseits das durch ACE2 aus Ang I und Ang II entstehende Ang-(1–7), das seine Hauptwirkung (vasodilatatorisch, antiproliferativ, antifibrotisch, antithrombotisch) hauptsächlich über den Mas-Rezeptor bewerkstelligt. Mit den RAS-Blockern beeinflussen wir einen Arm des Systems, ohne zu wissen, was am anderen Arm geschieht. Wahrscheinlich wird es in absehbarer Zeit möglich sein, mit Agonisten und Antagonisten in das Ang-(1–7)-Mas-Rezeptor- System einzugreifen und damit arterielle Hypertonie, chronische Herzinsuffizienz, hepatorenales Syndrom, diabetische Nephropathie und die Progression der chronischen Niereninsuffizienz zu beeinflussen.

Ursprünglich erschienen:
Neph 04|2011
Neph 04|2011