


























































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Studien aus 2018 bis 2019: Neues zur Glomerulonephritis
31. Mai 2019
Der folgende Artikel fasst eine Auswahl an Studien des vergangenen Jahres auf dem Gebiet der Glomerulonephritis zusammen, die (aus Sicht des Autors) bereits heute von Bedeutung für die klinische Tätigkeit sind oder Neuigkeiten beinhalten, die unser Krankheitsverständnis erweitern und in den nächsten Jahren zu Änderungen unserer diagnostischen oder klinischen Praxis führen könnten.
Anti-GBM-Erkrankung
Peroxidasin-Antikörper bei der Anti-GBM-Erkrankung (Goodpasture-Erkrankung): Die Basalmembran ist aus einem polymeren Netzwerk von Laminin und Kollagen Typ IV, die über extrazelluläre Matrixproteine miteinander verbunden sind, aufgebaut. Kollagen Typ IV setzt sich aus Triple-Helix-Protomeren zusammen, die aus drei verschiedenen α-Ketten bestehen. In der glomerulären und alveolären Basalmembran sind dies α3-, α4- und α5-Ketten. Die Triplehelices bilden ein zweidimensionales Netzwerk über End-zu-End-Verbindungen ihrer C-terminalen sogenannten nichtkollagenen Domänen (NC1), sodass Hexamere entstehen. Diese Hexamerstruktur wird von Schwefel-Stickstoff-Doppelbindungen enthaltenden Sulfiliminbrücken stabilisiert. Nur wenn diese Hexamere dissoziieren, können die pathogenen Epitope der NC1-Domäne der α3- und α5-Kette frei liegen und eine Autoantikörperbildung mit konsekutiver Entzündungsreaktion induzieren.1 Für die Bildung der stabilisierenden Sulfiliminbrücken ist das Enzym Peroxidasin (aus der Familie der Hämperoxidasen) verantwortlich. Bei 46 % der Goodpasture-Patienten in der Vanderbilt Goodpasture Cohort (n = 24) konnten zum Zeitpunkt der Erkrankung Autoantikörper gegen dieses Enzym nachgewiesen werden. Bei einem Drittel der Kohorte fanden sich auch gegen Myeloperoxidase (MPO) gerichtete Antikörper. Interessanterweise ließen sich Anti-Peroxidasin-Antikörper bei der Hälfte von Patienten mit Langzeitblutproben (n = 6) bereits Jahre vor der klinischen Erstmanifestation nachweisen (wie auch Anti-MPO- und Anti-GBM-Antikörper!). Von Patienten isolierte Anti-Peroxidasin-Antikörper hemmen in vitro Peroxidasin. Bei sogenannten „doppelt positiven“ Goodpasture-Patienten (Anti-GBM und Anti-MPO-Antikörper) ließ sich mittels kompetitiver ELISA nachweisen, dass bei einem Teil der Patienten die MPO-Reaktivität nur auf einer Kreuzreaktivität der Anti-Peroxidasin-Antikörper mit MPO (ebenfalls aus der Hämperoxidasefamilie mit Homologien zu Peroxidasin) beruht, während bei Anti-Peroxidasin-Antikörper-negativen doppelt positiven Goodpasture-Patienten die nachweisbaren Anti-MPO-Antikörper spezifisch gegen MPO gerichtet sind. Die klinische Implikation dieser Entdeckung ist, dass offensichtlich ein Teil der doppelt positiven Goodpasture-Patienten infolge einer Kreuzreaktivität falsch deklariert wird und somit keine (Vaskulitis-)Erhaltungstherapie benötigt. Auch bei 14 % von MPO-ANCA-Vaskulitis-Patienten konnten Anti-Peroxidasin-Antikörper nachgewiesen werden und waren mit einer höheren Krankheitsaktivität im Sinne eines höheren BVAS assoziiert.2 Offen bleibt, warum diese Anti-Peroxidasin-Antikörper entstehen und was ihre pathogenetische Bedeutung ist.
Fibrilläre Glomerulonephritis
DNAJB9 – neuer Biomarker für die fibrilläre Glomerulonephritis: Die fibrilläre Glomerulonephritis ist eine seltene glomeruläre Erkrankung, die sich klinisch mit Hämaturie, Proteinurie, Nierenfunktionseinschränkung und Hypertonie manifestiert, meist idiopathisch auftritt (in einem Drittel der Fälle assoziiert mit Hepatitis-C-Infektion, Paraproteinämie oder Autoimmunerkrankung) und mit einer schlechten renalen Prognose vergesellschaftet ist (50 % der Patienten werden innerhalb von 4 Jahren nach Diagnose dialysepflichtig). Die Diagnose beruht bislang auf der Zusammenschau der lichtmikroskopischen (Kongorot-negative mesangiale Expansion, mesangiale Hyperzellularität), immunfluoreszenzmikroskopischen (Nachweis von IgG und C3) und elektronenmikroskopischen (Nachweis willkürlich angeordneter Fibrillen mit 10–30 nm Durchmesser im Mesangium und/oder entlang der GBM) Befunde der Nierenbiospie. Mittels LC/MS konnte nun das Protein DNAJB9 (DNAJ Heat Shock Protein Family [Hsp40] Member B9) hochspezifisch in den Glomeruli von Patienten mit fibrillärer Glomerulonephritis überexprimiert nachgewiesen werden. Weder bei Patienten mit Amyloidose noch bei anderen Glomerulonephritiden wurde DNAJB9 exprimiert.3 Möglicherweise wird der Nachweis von DNAJB9, der auch einfach immunhistochemisch gelingt, Eingang in die Diagnosekriterien der fibrillären Glomerulonephritis finden und so die Diagnose dieser seltenen Erkrankung erleichtern.
Membranöse Glomerulonephritis
Nicht-invasive Diagnose der primären membranösen Glomerulonephritis: Seit der Entdeckung der M-Typ-Phospholipase-A2-Rezeptor-Antikörper (PLA2R-AK) stellt sich die Frage, ob in bestimmten Situationen oder bei bestimmten Patienten die Diagnose einer primären membranösen Glomerulonephritis (GN) alleinig nicht-invasiv mit dem Nachweis positiver PLA2R-AK und Verzicht auf Nierenbiopsie gestellt werden kann. Bobart und Kollegen haben versucht, diese Frage in einer retrospektiven Analyse aller an den drei Mayo-Kliniken zwischen 2015 bis 2018 auf PLA2R-AK getesteten Patienten (n = 838) zu beantworten. Bei 132 PLA2R-AK-positiven Patienten war auch eine Nativnierenbiopsie durchgeführt worden. Für eine positive PLA2R-Serologie mussten sowohl im Immunfloureszenzassay (indirekter Immunfluoreszenztest) ein positiver Antikörpernachweis als auch im ELISA (Euroimmun Assay) eine Antikörperkonzentration ≥ 2 RE/ml vorliegen. Nach klinischem, laborchemischem oder bildgebendem Ausschluss sekundärer Ursachen blieben 97 Patienten mit positiver Serologie und Nierenbiopsie für die Analyse übrig, die abhängig von der eGFR in 2 Gruppen unterteilt wurden: Patienten mit erhaltener Nierenfunktion (eGFR ≥ 60 ml/min, n = 60) und solche mit eingeschränkter Nierenfunktion (eGFR < 60 ml/min, n = 37). Bei 58 der 60 Patienten mit erhaltener Nierenfunktion fand sich histologisch nur eine membranöse GN. Die bei einem Patienten nachgewiesenen FSGS-Läsionen und die mit einer diabetischen Nierenerkrankung zu vereinbarenden Befunde beim anderen zogen keine weiteren diagnostischen oder therapeutischen Konsequenzen nach sich. Demgegenüber ließen sich bei 5 der 37 Patienten mit eGFR < 60 ml/min (13,5 %) histologisch neben der membranösen GN noch weitere, teils differenzialdiagnostisch und therapeutisch relevante Zweitbefunde nachweisen (z. B. akute interstitielle Nephritis, proliferative GN mit Crescents). Die Hälfte aller PLA2R-AK-negativen Patienten (180/348, sekundäre Formen inklusive) zeigten im Biopsat eine membranöse GN. Basierend auf diesen Ergebnissen schlagen die Autoren den in der Abbildung dargestellten Diagnosealgorithmus vor.4
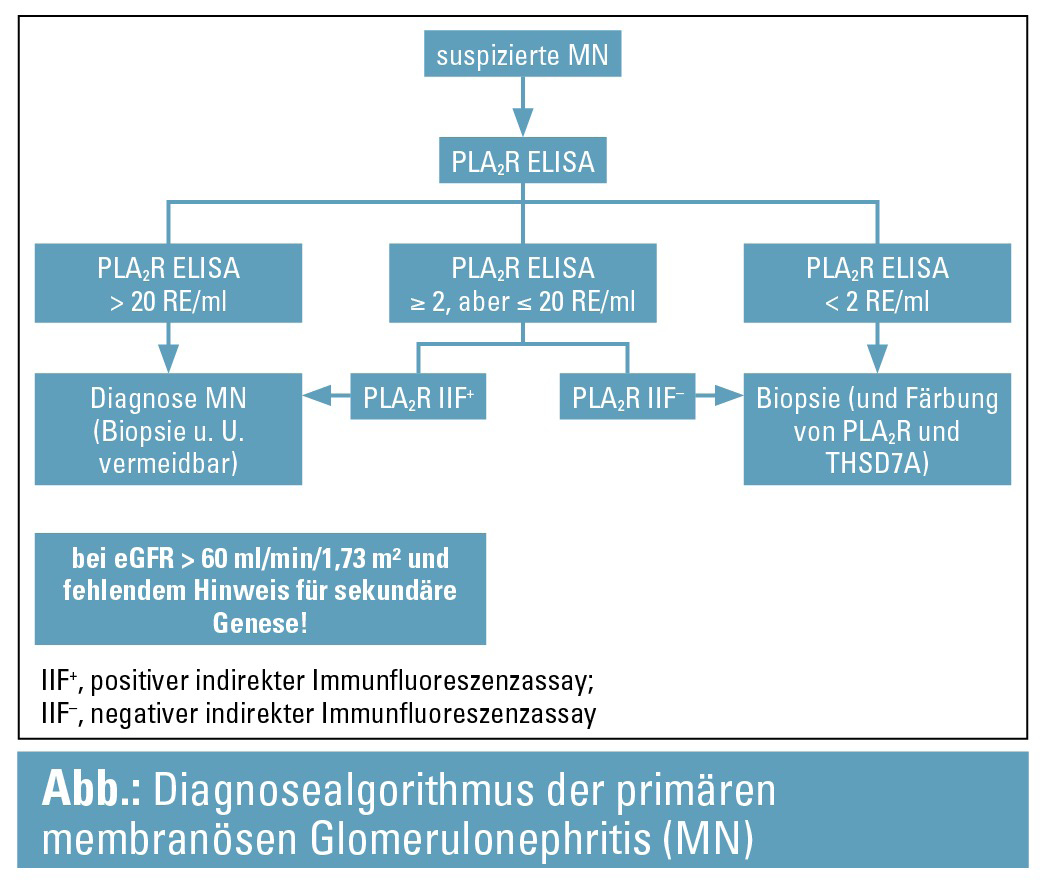
Globale Glomerulosklerose
Klinische Bedeutung der globalen Glomerulosklerose – Einfluss der Altersadjustierung: Nicht selten finden sich in einer Nierenbiopsie ein oder mehrere global sklerosierte Glomeruli. Diese können sowohl im Rahmen des normalen Alterungsprozesses als auch als Folge (und Endzustand) glomerulärer Erkrankungen auftreten. Abhängig von der Anzahl der in der Biopsie erfassten Glomeruli wurden altersabhängige obere Grenzwerte für global sklerosierte Glomeruli festgelegt (https://qxmd.com/calculate/glomerulosclerosis-on-biopsy-2015).5
Haben global sklerosierte Glomeruli nun per se eine klinische Bedeutung hinsichtlich der renalen Prognose oder nur, wenn ihre Zahl den altersabhängigen Grenzwert überschreitet? Anhand von Biopsie- und klinischen Outcomedaten von 425 Patienten mit proteinurischer Nierenerkrankung (Minimal Change Disease, FSGS, membranöse GN) fand sich bei Patienten mit über der Altersnorm liegender globaler Glomerulosklerose (n = 148) eine signifikant höhere CKD-Progressionsrate (definiert als ESKD oder eGFR-Abfall > 40 %) über 15 Jahre als bei Patienten mit altersentsprechender globaler Glomerulosklerose (n = 107) oder Patienten ohne globaler Glomerulosklerose (n = 170). Auch nach Adjustierung auf histologische (interstitielle Fibrose, Aterio- und Arteriolosklerose), klinische (Hypertonie, Diabetes mellitus, Body-Mass-Index, Alter, renale Grunderkrankung) und renale (eGFR, Proteinurie) Einflussgrößen blieb das Progressionsrisiko noch um 80 % erhöht.6 Nach Jahrzehnten der Fokussierung auf das Tubulointerstitium (interstitielle Fibrose, Tubulusatrophie) als dem entscheidenden Nephronkompartiment für die renale Langzeitprognose rückt diese Studie auch das Glomerulum wieder etwas ins Scheinwerferlicht.
Fokal segmentale Glomerulosklerose
Sparsentan bei primärer FSGS: Aus präklinischen Studien wissen wir, dass neben Angiotensin II auch erhöhtes Endothelin-1 neben verschiedenen renalen Effekten podozytenschädigende Wirkung hat. In FSGS-Tiermodellen und bei Patienten mit diabetischer Nierenerkrankung konnte für Endothelinrezeptorantagonisten ein zusätzlicher antiproteinurischer Effekt gezeigt werden, wenn sie mit RAS-Blockern kombiniert wurden.7, 8 In einer 8 Wochen dauernden doppelblinden, kontrollierten Phase-II-Studie (DUET) wurde nun der erste oral wirksame duale Endothelin-1-Typ-A-Rezeptor-(ETA)- und Angiotensinrezeptorblocker Sparsentan (3 Dosierungsgruppen) im direkten Vergleich mit Irbesartan (300 mg/Tag) bei Patienten mit bioptisch oder genetisch gesicherter primärer FSGS (n =109, eGFR > 30 ml/min, Eiweiß-Kreatinin-Quotient ≥ 1,0 g/g) überprüft. Primärer Endpunkt war die Änderung der Proteinurie nach 8 Wochen, sekundärer Endpunkt die Zahl der Patienten mit partieller Remission (Proteinurie ≤ 1,5 g/g und > 40 % Reduktion der Proteinurie). In der gepoolten Analyse fand sich bei den mit Sparsentan behandelten Patienten eine stärkere Reduktion der Proteinurie (–44,8 vs. –18,5 %, p = 0,006) und ein höherer Anteil an Patienten mit partieller Remission (28,1 vs. 9,4 %, p = 0,040). Die Blutdrucksenkung unter Sparsentan war etwas stärker, beim eGFR-Verlauf zeigte sich in dieser kurzen Zeit kein Unterschied. Die Verträglichkeit war gut, auch hinsichtlich der bislang bei Endothelinrezeptorantagonisten besorgniserregenden Nebenwirkung der Flüssigkeitsretention waren in dieser zugegebenermaßen kurzen Beobachtungszeit keine kritischen Sicherheitssignale zu erkennen.9 Die entsprechende Phase-III-Studie (DUPLEX) mit 2-Jahres-Daten ist bereits initiiert.
Minimal Change Disease
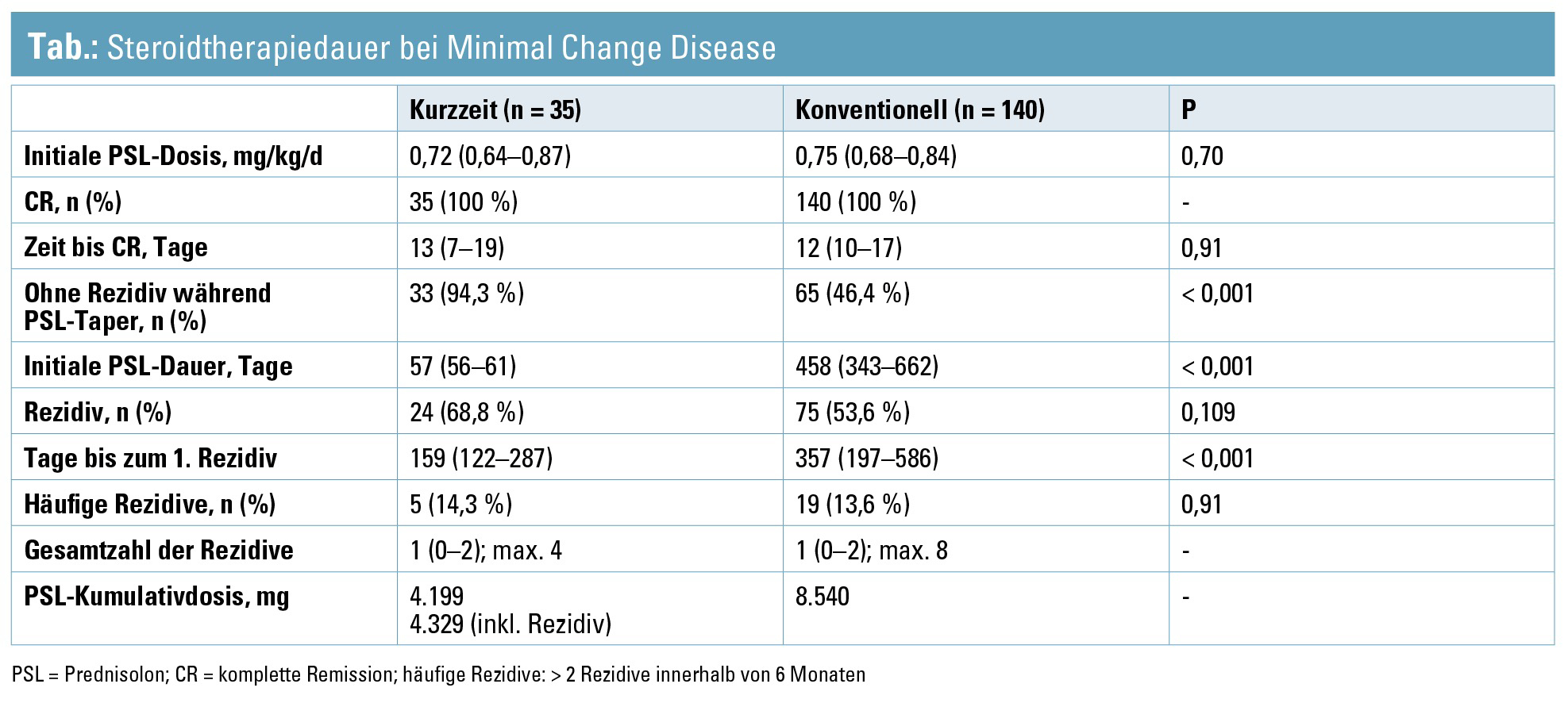
Dauer der Steroidtherapie bei Minimal Change Disease – in der Kürze liegt die Würze: In den (noch) gültigen KDIGO-Glomerulonephritis-Richtlinien von 2012 wird bei der Minimal Change Disease (MCD) des Erwachsenen eine Steroidtherapiedauer von bis zu einem halben Jahr empfohlen. In einer rezenten japanischen prospektiven Multicenterbeobachtungsstudie wurde nun über die erfolgreiche Anwendung eines Kurzzeitsteroidregimes über 2 Monate berichtet. Insgesamt 35 Patienten wurden mit dem kurzen Regime (Prednisolon 0,8–1,0 mg/kg/Tag – max. 60 mg für 4–6 Wochen, dann Reduktion auf 2/3 der Initialdosis für weitere 4 Wochen) behandelt und mit einer historischen MCD-Kohorte von 140 Patienten (Prednisolon 0,8–1,0 mg/kg/Tag – max. 60 mg für 2–4 Wochen, dann Reduktion um 5–10 mg alle 2 bis 4 Wochen, 5–10 mg Erhaltungstherapie für [1 bis] 2 Jahre zur Rezidivprophylaxe) verglichen. Alle Patienten litten an einer bioptisch gesicherten MCD mit Erstepisode eines nephrotischen Syndroms und alle erreichten nach 4 Wochen Therapie eine komplette Remission (CR). Initiale Prednisolondosis, CR-Rate und Dauer bis zur CR waren ident, die Dauer der initialen Prednisolontherapie und die kumulative Steroiddosis unterschieden sich jedoch dramatisch (Tab.). Die Rezidivrate war in der Kurzzeitgruppe numerisch höher, die Dauer bis zum ersten Rezidiv signifikant kürzer, die Zahl der Patienten mit häufigen Rezidiven jedoch vergleichbar. Bei fehlender statistischer Signifikanz erkrankten in der konventionellen Gruppe numerisch doch deutlich mehr Patienten an Diabetes mellitus (11,4 vs. 2,9 %).10
AUSBLICK: Nach diesem wieder erkenntnisreichen Glomerulonephritis-Jahr 2018–2019 dürfen wir schon sehr gespannt sein auf die neuen KDIGO-Richtlinien, die derzeit gerade in Überarbeitung sind und vielleicht schon kommendes Jahr vorgestellt werden können.
1 Pedchenko V., NEJM 2010
2 McCall A.S., JASN 2018
3 Dasari S., JASN 2018
4 Bobart S.A., Kidney Int 2019
5 Kremers W.K., NDT 2015
6 Hommos M.S., Kidney Int 2018
7 Kohan D.E., Kidney Int 2014
8 Cherney D.Z.I., Kidney Int Suppl 2018
9 Trachtman H., JASN 2018
10 Ozeki T., Am J Nephrol 2019

OA Priv.-Doz. Dr. Emanuel Zitt
Innere Medizin III (Nephrologie und Dialyse), Akademisches Lehrkrankenhaus LKH Feldkirch

Ursprünglich erschienen:
Neph 02|2019
Neph 02|2019
Herausgeber: Österreichische Gesellschaft für Nephrologie, Univ.-Prof. Dr. Rainer Oberbauer, Klinische Abteilung für Nephrologie und Dialyse, Universitätsklinik für Innere Medizin III, Medizinische Universität Wien
Publikationsdatum: 2019-05-31
Zur Ausgabe »
Publikationsdatum: 2019-05-31
Zur Ausgabe »