


























































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
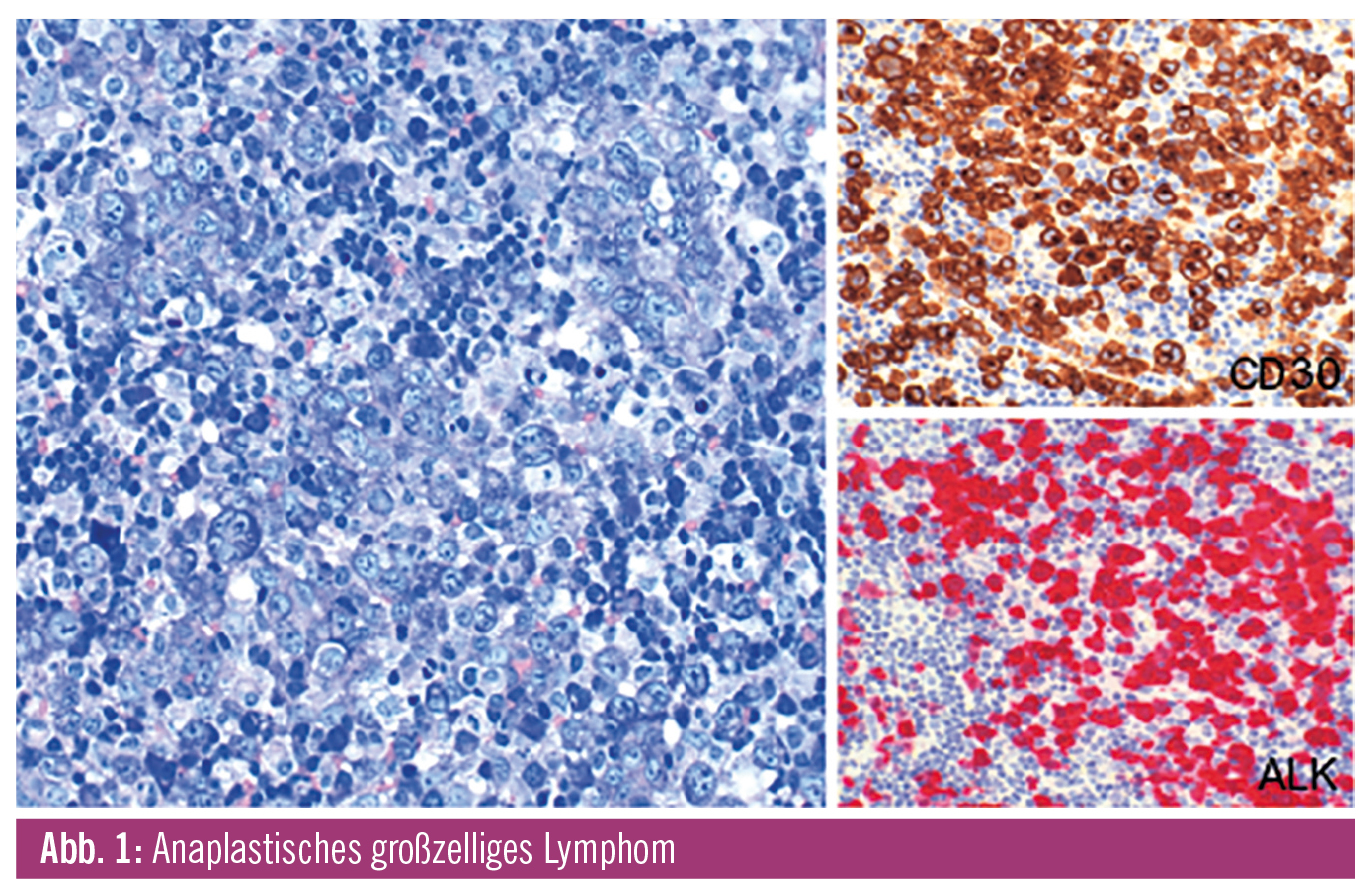
Anaplastisches großzelliges Lymphom
20. September 2018
Das anaplastische großzellige Lymphom (ALCL) wurde erstmals 1985 von Harald Stein beschrieben und ist ein seltenes aggressives, CD30-positives Non-Hodgkin-Lymphom (NHL). Die Klonierung von ALK im Jahr 1994 war bahnbrechend für weitere Studien in der Aufklärung von Mechanismen, die das ALK-Onkogen in der Lymphomentstehung beisteuert. Basierend auf der ALK-Expression werden zwei Subklassen von ALCL definiert, welche signifikant unterschiedliche klinisch-pathologische Eigenschaften aufweisen.
Epidemiologie
ALCL treten gehäuft im Kindes- und frühen Erwachsenenalter auf und machen in dieser Population 10–15 % aller Lymphome aus; im Erwachsenenalter kommen sie hingegen selten vor (ca. 1–2 % der NHL). Bei Kindern zeigen > 90 % der Fälle eine Translokation des ALK-Gens auf Chromosom 2p23, wobei als häufigster Translokationspartner Nucleophosmin (NPM) auf Chromosom 5q35 fungiert. Im Erwachsenenalter sind nur ca. 40–50 % der Fälle mit einem ALK-Rearrangement assoziiert.
Die systemischen ALK-rearrangierten Lymphome werden aufgrund ihrer klinisch-pathologischen Charakteristika in einer eigenständigen Gruppe als „ALK+ ALCL“ von ALK-negativen ALCL und zusätzlich von der Gruppe der primär kutanen ALCL (cALCL) abgegrenzt. Letztere sind in fast allen Fällen ALK-negativ und präsentieren sich als lokalisierte oder solitäre, häufig ulzerierte Hauttumoren mit zumeist günstiger Prognose. Eine neue provisorische Entität stellen äußerst selten beschriebene „Brustimplantat“-assoziierte ALCL (biALCL) dar.
Klinische Charakteristika
Systemische ALCL werden klinisch oft erst in einem höheren Stadium (III und IV) diagnostiziert und präsentieren sich mit häufig auftretender B-Symptomatik. Extranodale Infiltrate (Haut, Leber, Lunge, Weichteile, Knochen) sind in vielen Fällen bereits von Anfang an vorhanden. Eine Knochenmarkinfiltration ist selten (ca. 15 %), häufig sehr diskret ausgeprägt und wird manchmal erst immunhistologisch detektiert.
Morphologie
ALCL zeigen typischerweise flächige Verbände von großen Tumorzellen, wobei eine sinusoidale Ausbreitung mit perivaskulärer Akzentuierung charakteristisch ist. Zytologisch weisen ALK+ ALCL eine heterogene zelluläre Zusammensetzung auf: Etwa 60 % der Fälle zeigen einen reinen „Common“-Subtyp mit kohäsiv gelagerten Tumorzellen, welche exzentrische, teils nierenförmige Zellkerne besitzen; zusätzlich zeigen sie eine typische paranukleäre eosinophile Aufhellung. Diese Zellen sind sogenannte „Hallmark Cells“ und sollten in allen morphologischen Varianten identifiziert werden können (Abb. 1). Die morphologischen Varianten inkludieren einen kleinzelligen, einen lymphohistiozytären und einen Hodgkin-artigen Typ. Die kleinzellige Variante (5–10 %) ist zumeist durch kleine, monomorphe Zellen charakterisiert. Diese Variante ist die am häufigsten als peripheres T-Zell-Lymphom (PTCL) fehldiagnostizierte Variante. In ca. 20 % der Fälle zeigt sich eine Kombination aus mehreren unterschiedlichen Mustern.
ALK-negative ALCL sind rein morphologisch nicht von ALK+ ALCL zu unterscheiden, jedoch sind morphologische Varianten selten; eine kleinzellige Variante kommt definitionsgemäß nicht vor.
Immunphänotyp und Genetik
Immunmorphologisch sind die Tumorzellen neben der starken Expression von CD30 (Abb. 1) durch einen inkompletten T-Zell-Immunphänotyp mit häufiger Expression zytotoxischer Antigene wie TIA-1, Granzyme B und Perforin charakterisiert. Manche Fälle werden aufgrund des Fehlens von Pan-T-Zellmarkern (z. B. CD2, CD3, CD4, CD5, CD8, T-Zell-Rezeptor) als „Null-Zell-Typ“ bezeichnet, deren Herkunft von T-Zellen sich erst auf genetischem Level beweisen lässt. Die Ursprungszelle von ALCL wurde in einer zytotoxischen peripheren T-Zelle vermutet; rezent wurde jedoch eine Gensignatur in ALCL-Zellen detektiert, welche charakteristisch für thymische Vorläuferzellen ist. Es wird daher spekuliert, dass die ALK-Translokation bereits in Vorläuferzellen des Thymus auftritt und sekundäre genetische Events in zirkulierenden ALK+-Thymozyten zur Entstehung von ALCL führen.
Das immunhistologisch darstellbare ALK-Protein wird von den Tumorzellen im Fall der am häufigsten zugrunde liegenden chromosomalen Translokation zwischen ALK und NPM-1 sowohl nukleär/nukleolär als auch zytoplasmatisch exprimiert (Abb. 1). Interessanterweise findet man in vielen Fällen der kleinzelligen Variante eine ausschließlich nukleäre Expression, während die großen perivaskulären Hallmark-Zellen in diesen Fällen zusätzlich eine zytoplasmatische Reaktivität zeigen. Infolge von Variant-Translokationen mit anderen Bindungspartnern als NPM-1 (z. B. TPM3, ATIC, TFG, CLTC, MSN oder TPM4) ist die ALK-Expression ausschließlich zytoplasmatisch oder membranös nachweisbar. Die Fusion dieser Gene führt zu einer Hinaufregulation von ALK und einer Aktivierung der Kinase-Funktion. Die bessere Prognose der ALK+-Fälle gegenüber ALK-negativen Fällen ist unabhängig vom jeweiligen Fusionspartner.
Etwa 30 % der ALK-negativen Fälle sind mit einem Rearrangement von DUSP22-IRF4 assoziiert, weitere 8 % zeigen ein TP63-Rearrangement. DUSP22-Rearrangements wurden auch in Fällen von cALCL sowie in Fällen von lymphomatoider Papulose beschrieben.
Die DUSP22+-Fälle sind morphologisch durch flächige Verbände von „monomorphen“ Hallmark-Zellen charakterisiert und exprimieren häufig keine zytotoxischen Marker. Der Nachweis einer DUSP22-Assoziation in ALK-negativen Fällen ist von klinisch-prognostischer Relevanz, DUSP22+-Fälle zeigen nämlich eine ähnlich gute Prognose wie ALK+ ALCL. Im Gegensatz dazu weisen Fälle mit TP63-Rearrangement einen aggressiven Verlauf mit schlechter Prognose auf. Die triple-negativen ALCL (ALK/DUSP22/TP63) zeigen eine intermediäre Prognose.
Zur prognostischen Abklärung der verschiedenen ALCL-Formen empfiehlt sich eine algorithmische diagnostische Aufarbeitung, wie in Abbildung 2 dargestellt: Die immunhistologische Färbung mit p63-Antikörpern ist zwar sensitiv auf das Vorliegen einer TP63-Translokation, aber nicht spezifisch. Der Nachweis eines TP63-Rearrangements ist nicht spezifisch für systemische ALCL, sondern wurde selten auch in primären cALCL (aggressive Verlaufsform), in PTCL und in einzelnen DLBCL beschrieben. cALCL sollten klinisch unbedingt von systemischen ALCL abgegrenzt werden. Etwa die Hälfte der ALK-negativen ALCL weist – analog zu ALK+ ALCL – eine Aktivierung von STAT3 auf und zahlreiche Tumoren zeigen Mutationen in JAK-1 und STAT3. Ferner wurden auch Translokationen mit ROS1, TYK2 und FRK beschrieben. Die Vielzahl an genetischen Events, welche letztlich allesamt zu einer Aktivierung des JAK-STAT3-Pathways führen, macht ALK-negative ALCL zu therapeutischen Kandidaten für JAK-STAT3-Inhibitor-Therapeutika.
Die biALCL stellen eine diagnostische Rarität dar. Sie sind häufig mit Seromen oder Kapselbildungen, die um das Implantat auftreten können, assoziiert. Das Vorkommen einer tumorösen Masse stellt einen negativen Prognosefaktor dar. biALCL entsprechen morphologisch und immunmorphologisch den anderen ALK-negativen Lymphomen. Eine Assoziation mit DUSP22 und TP63 wurde bisher nicht gefunden, Mutationen von JAK-1 und STAT1 wurden hingegen beschrieben.

Resümee
Prognostisch wichtige Rearrangements von ALK, DUSP22 und TP63 können mittels IHC und FISH nachgewiesen werden. Die Aufklärung genetischer Events mit Detektion von Non-ALK-Kinase-Genen in ALK-negativen ALCL, welche zu einer Aktivierung von STAT3 führen, erlaubt zukünftig innovative Therapieansätze in den klinisch bisher teilweise ungünstig verlaufenden ALK-negativen ALCL.
Ausgewählte Literatur:
– Benharroch D et al., Blood 1998; 91(6):2076–84
– Falini B et al., Br J Haematol 2001; 114(4):741–60
– Hapgood G, Savage KJ, Blood 2015; 126(1):17–25
– Laurent C et al., Leukemia 2012; 26(1):188–90
– Morris SW et al., Science 1994; 263(5151):1281–84
– Parrilla Castellar ER et al., Blood 2014; 124(9):1473–80
– Stein H et al., Blood 2000; 96(12):3681–95
– Story SK et al., Oncologist 2013; 18(3):301–07
– Vasmatzis G et al., Blood 2012; 120(11):2280–89

AutorIn: Univ.-Prof. Dr. Ingrid Simonitsch-Klupp
Institut für Klinische Pathologie,Medizinische Universität Wien, AKH Wien

Ursprünglich erschienen:
Patho 01|2018
Patho 01|2018