


























































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Revision der 4. Auflage der WHO-Klassifikation: Klinisch relevante Neuerungen
20. September 2018
Die WHO-Klassifikation maligner Tumoren des blutbildenden Systems ist als großer Fortschritt in der Therapie und Diagnose zu sehen, insbesondere auf dem Gebiet der lymphatischen Neoplasien. Nach Jahrzehnten einer länder- bzw. gebietsspezifischen Lymphom-Diagnostik (Klassifikationsschemata nach Rappaport, Lukes & Collins, Working Formulation, Kieler Klassifikation, REAL)1 war die Veröffentlichung der WHO-Klassifikation 2001 ein Meilenstein. Denn erstmals wurde ein Lymphom sowohl in Amerika als auch in Europa und in Japan gleich diagnostiziert und beurteilt. Die nunmehrige revidierte 4. Auflage dieser WHO-Klassifikation2, 3 birgt einige Neuerungen und Präzisierungen. Es wurden sowohl frühe Formen von Lymphomen und Grenzen zu reaktiven Veränderungen exakter definiert als auch hochaggressive Lymphome unter Einbeziehung klinischer und molekularpathologischer Daten neu geordnet. Dieser Artikel soll einen kurzen Überblick über klinisch relevante Neuerungen geben.
Neuerungen
Nachdem es sich bei der aktuellen WHO-Klassifikation um eine revidierte Fassung der 4. Auflage und nicht um eine komplett neu gestaltete 5. Auflage handelt, beinhaltet diese keine neuen definitiven Entitäten. Es wurden lediglich einige wenige provisorische Entitäten aufgenommen und einige wenige bislang provisorische Entitäten zu definitiven Entitäten erklärt, wie beispielsweise das pädiatrische follikuläre Lymphom (PTFL). Diese Unterscheidung ist jedoch im klinischen Alltag kaum relevant, lediglich die Kodierung nach ICD-O ist definitiven Entitäten vorbehalten (Tab.).
Diese Zusammenstellung verdeutlicht die wesentliche Ausrichtung der revidierten Fassung: eine exaktere Definition insbesondere der Grenzbereiche zwischen „klassischem Lymphom“ und zum Teil aggressiv fortschreitenden, teils indolenten reaktiven Veränderungen. War das PTFL in der ersten Auflage noch als Variante des follikulären Lymphoms angeführt, hat sich seither gezeigt, dass diese Entität nicht nur bei Kindern und Jugendlichen zu beobachten ist, sondern dass dieses – meist in Halslymphknoten isoliert auftretende Lymphom mit charakteristischen landkarten- oder puzzleartigen großen „Keimzentren“ mit zahlreichen blastoiden Zellen und auffallend breiten Mantelzonen – nur schwer von einem follikulären Lymphom Grad 3A oder 3B abzugrenzen ist. BCL2- oder BCL6-Rearrangements sind definitionsgemäß auszuschließen, für die Diagnostik wird eine Untersuchung auf Immunglobulin-Leicht- oder -Schwerketten-Rearrangement gefordert. Grund für die Neueinordnung ist aber, dass dieser Typ eines Keimzentrumslymphoms mit spezifischer Mutation eine exzellente Prognose aufweist und mittlerweile nicht mehr chemotherapeutisch behandelt, sondern engmaschig kontrolliert wird.4
Auch die Abgrenzung plasmazellreicher Varianten kleinzelliger B-Lymphome, lymphoplasmazytisches Lymphom und Marginalzonenlymphom mit dem geforderten Nachweis einer MYD88-L265P-Mutation soll nunmehr für eine klarere Abgrenzung dieser beiden Entitäten sorgen. Bisher zeigten Marginalzonenlymphome in zahlreichen Studien sowohl im klinischen Verlauf als auch im Phänotyp hohe Heterogenität, die nun durch die klare molekularpathologische Abgrenzung des lymphoplasmazytischen Lymphoms (mit nachgewiesener MYD88-L265P-Translokation) reduziert wird.

„Hochmaligne“ B-Zell-Lymphome
Die größten Änderungen dieser revidierten 4. Fassung der WHO-Klassifikation betreffen aggressive B-Zell-Lymphome. Diese machen in Europa rund ein Drittel aller Lymphome aus – einerseits die diffusen großzelligen B-Zell-Lymphome (DLBCL), andererseits die High-Grade-B-Zell-Lymphome (HGBL). Letztere sind ein neuer Terminus für Lymphome, die bisher als sogenanntes „Grauzonen-Lymphom“ oder als „B-Zell-Lymphom, unklassifiziert mit Eigenschaften zwischen einem DLBCL und einem Burkitt-Lymphom“ bezeichnet wurden. Eine Übertragung des Terminus „high-grade“ als „hochmaligne“ soll vermieden werden, da dieser Terminus noch aus Zeiten der Kiel-Klassifikation geläufig ist, damals jedoch völlig anders definiert war.
Gefordert („Mandatory for Diagnosis“) für eine exakte Diagnostik wird nunmehr:
- Angabe des molekularpathologischen Typs der Ursprungszelle (COO – cell of origin): Stammt das Lymphom von aktivierten B-Zellen (ABC – activated B-cell) oder von Keimzentrumszellen (GCB – germinal center B-cell) ab?
- Liegen spezifische Assoziationen mit einem infektiösen Agens (EBV-positives DLBCL, EBV-assoziiertes mukokutanes Ulkus, HHV-8-positives DLBCL, HHV-8-positive germinotrope lymphoproliferative Erkrankung) oder eine das Immunsystem chronisch modulierende Situation (Implantat-assoziiertes anaplastisches großzelliges Lymphom, DLBCL assoziiert mit chronischer Entzündung) vor?
- Liegen spezifische genetische Aberrationen (IRF4, ALK) vor?
Die Angabe des Typs der COO hat derzeit (noch?) keine Auswirkungen, weder auf die Therapie noch auf die Prognose – begründet unter anderem darin, dass immunhistochemisch vor allem nur bestimmt werden kann, ob eine Abstammung von GCB vorliegt oder nicht (Non-GCB)5; ein rein immunhistochemischer Beweis des ABC-Typs ist derzeit noch nicht möglich und diverse Algorithmen (Hans, Choi, Visco-Young, …) weisen unterschiedlich hohe (maximal 87 %) Konkordanzen mit den Ergebnissen genetischer Untersuchungen auf. Studien zur klinischen Relevanz dieser Unterscheidung zeigten zum Teil beträchtliche prognostische Unterschiede, andere Studien wiederum konnten diese Unterschiede nicht nachweisen.6 Somit kann derzeit noch kein endgültiges Urteil über die Wertigkeit dieser, von der WHO als obligatorisch angesehenen Unterteilung getroffen werden.
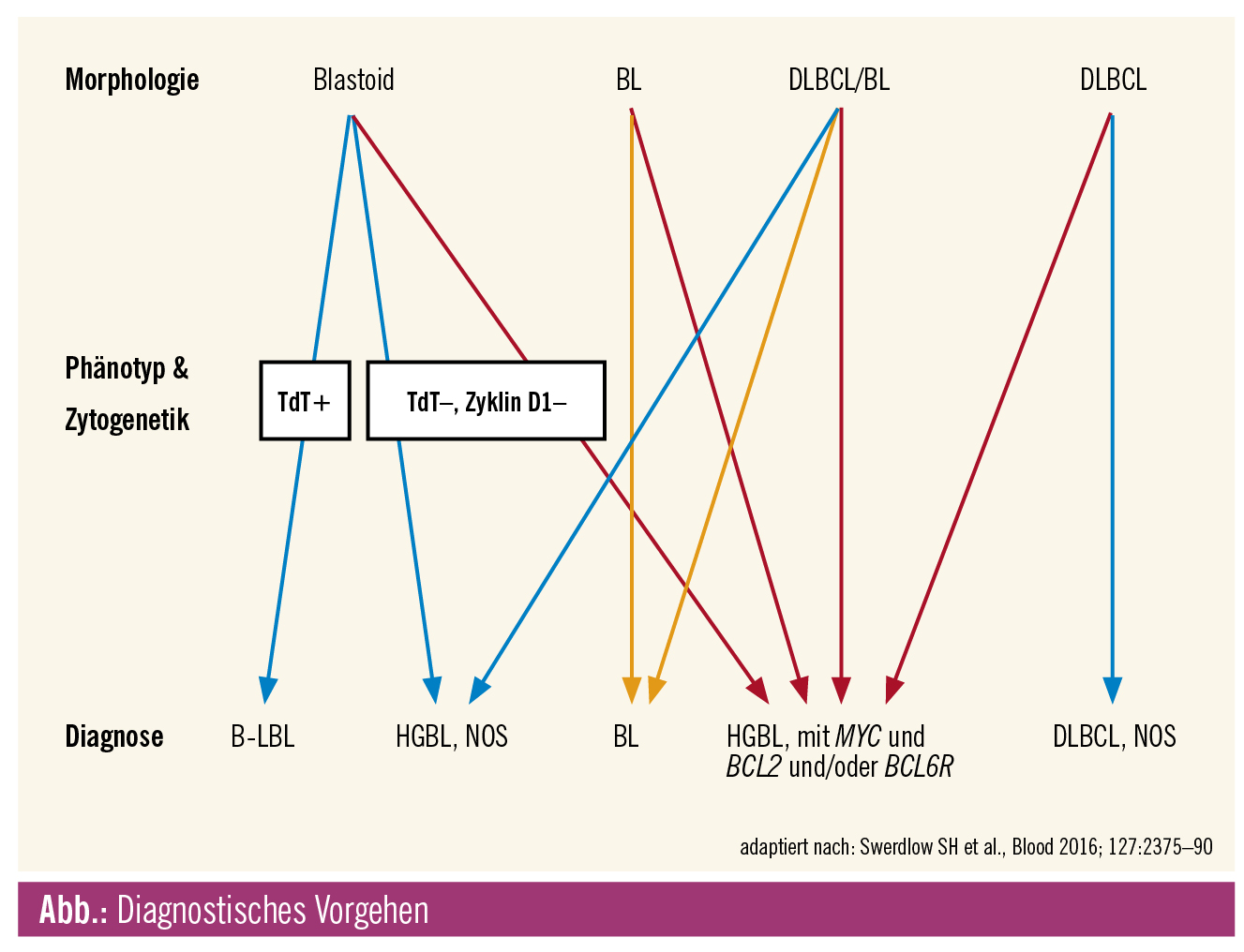
Bei diesen hochmalignen Lymphomen werden außerdem Typen mit MYC- und BCL2- und/oder BCL6-Translokation (sog. Double-Hit- oder Triple-Hit-Lymphome) unterschieden – nunmehr sind diese Untersuchungen auch bei klassischen diffusen großzelligen B-Zell-Lymphomen mit typischer Morphologie (reichlich Centro- oder Immunoblasten, kein Sternhimmelbild) durchzuführen –, die dann derzeit trotz ihrer eindeutigen morphologischen Zuordnung zur neu geschaffenen Gruppe der High-Grade-B-Zell-Lymphome gezählt werden (Abb.). Der Nachweis einer Translokation des MYC-Gens sollte mit einer etablierten MYC-Break-Apart-Sonde mittels Fluoreszenz-in-situ-Hybridisierung (FISH) erfolgen. Ob ein immunhistochemisches „Screening“ mit einem Cut-off von 30 % positiven Zellen (vorausgesetzt, das Material ist optimal fixiert und die Zeit zwischen Gewebsentnahme und Fixierung ist so kurz wie möglich) die Anzahl dieser aufwendigen Untersuchung (weniger als 10 % aller DLBCL zeigen MYC-Translokationen) reduzieren kann, ist noch nicht endgültig entschieden. Das Panel der Referenzpathologen für Lymphome in Deutschland7 empfiehlt diese Untersuchung in ihrem Konsensus vom April 2018, da in bis zu 20 % der Fälle auch bei immunhistochemisch negativem MYC-Nachweis Translokationen gefunden werden konnten. Die eigenen Erfahrungen zeigen jedoch eine weit höhere Konkordanz zwischen Immunhistochemie und FISH sowie eine Diskordanz lediglich in wenigen Fällen mit suboptimaler Fixierung.
Bei der Einordnung aggressiver Lymphome in diese neue Gruppe sind folgende Voraussetzungen zu beachten:
- kein follikuläres Wachstum erkennbar
- reifzelliges B-Zell-Lymphom, d. h. kein Nachweis von Markern des Precursor-Typs (TdT-negativ)
- Ausschluss der blastoiden Variante eines Mantelzelllymphoms
- Ausschluss eines anderen spezifischen Subtyps
Resümee und Ausblick
Die Revision der 4. Auflage der WHO-Klassifikation weist einige, auch für Kliniker relevante Neuerungen auf und bildet neue (durch neue molekularpathologische Methoden) getriggerte Entitäten ab. Wichtige neue Informationen betreffen vor allem die Abgrenzung zu indolenten Vorstufen und reaktiven Prozessen sowie am anderen Ende die Neudefinition von hochaggressiven B-Zell-Lymphomen. Vor allem aber zeigt die revidierte Fassung die enorme Wichtigkeit der Kommunikation zwischen klinisch-therapeutisch und diagnostisch tätigen Kolleginnen und Kollegen: Die Angabe „Schwellung, Lymphom?“ ist für eine exakte Diagnostik nicht mehr ausreichend.
1 Lennert K: History of the European Association for Haematopathology. Springer Verlag, Berlin-Heidelberg 2006
2 Swerdlow SH et al. (Hrsg.): WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. International Agency for Research on Cancer; 4. Auflage, Lyon 2017
3 Swerdlow SH et al., Blood 2016; 127:2375–90
4 Louissaint Jr. A et al., Blood 2016; 128:1093–100
5 Ott G et al.: Revidierte Fassung der 4. Ausgabe der WHO-Klassifikation maligner Lymphome – was ist neu? Der Pathologe, Springer Medizin Verlag GmbH [ahead of print 17.7.2018]
6 Jaffe ES et al.: Understanding the New WHO Classification of Lymphoid Malignancies: Why It’s Important and How It Will Affect Practice. American Society of Clinical Oncology – Educational Book 2017; 37:535–46
7 Klapper W et al.: Aggressive B-Zell-Lymphome – Empfehlungen des Deutschen Panels der Referenzpathologen im Kompetenznetz Maligne Lymphome e.V. zum diagnostischen Vorgehen nach der aktuellen WHO-Klassifikation, Update 2017. Der Pathologe, Springer Medizin Verlag GmbH [ahead of print 17.4.2018]

AutorIn: Dr. Alexander Nader, MSc
Institut für Pathologie und Mikrobiologie,Ferdinand-Hanusch-Krankenhaus der WGKK, Wien

Ursprünglich erschienen:
Patho 01|2018
Patho 01|2018