


























































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Klassifizierung der Hodgkin-Lymphome
20. September 2018
Die makroskopische Erstbeschreibung des Hodgkin-Lymphoms erfolgte 1832 durch den Namensgeber Thomas Hodgkin. Die histologische Charakterisierung der typischen Tumorzellen erfolgte 1898 von Carl Sternberg (Wien) und 1902 von Dorothy Reed (USA).
Der Nachweis der typischen einkernigen Hodgkin-Zellen und der mehrkernigen Reed-Sternberg-Zellen (HRS) unterscheidet das Hodgkin-Lymphom von anderen Lymphomen (Non-Hodgkin-Lymphome) sowie Lymphknotenerkrankungen anderer Ursache, wie beispielsweise viral verursachten Lymphadenopathien, insbesondere durch das Epstein-Barr-Virus (EBV) verursachte Erkrankungen. Zumeist entsprechen mehr als 90 % der Zellen in Hodgkin-Lymphomen reaktiven Zellen mit variabler Anzahl von Lymphozyten, Plasmazellen, Histiozyten und Eosinophilen. Auch Nekrosen können vorkommen.
Im deutschsprachigen Raum beträgt die Inzidenz des Hodgkin-Lymphoms 2–3 Neuerkrankungen pro 100.000 Personen jährlich.
Klassifikation
Die aktuelle WHO-Klassifikation1 unterscheidet – basierend auf Morphologie und Immunphänotyp der Tumorzellen – 2 Haupttypen von Hodgkin-Lymphomen: das noduläre lymphozytenprädominante Hodgkin-Lymphom (NLPHL) und das wesentlich häufigere klassische Hodgkin-Lymphom (cHL). Bei Letzterem sind histologisch 4 Subtypen zu unterscheiden: nodulär sklerosierender Subtyp (NS), lymphozytenreicher Subtyp (LR), Mischtyp und der extrem seltene lymphozytenarme Subtyp.
NLPHL: Beim NLPHL handelt es sich um ein B-Zell-Lymphom, welches ca. 5 % aller Hodgkin-Lymphome ausmacht. Betroffen sind meist männliche Patienten zwischen 25 und 40 Jahren. Bevorzugt werden zervikale und inguinale Lymphknoten (LK) befallen. Zum Zeitpunkt der Diagnosestellung liegt meist ein frühes Stadium vor, ein krankheitsbedingter Tod ist extrem selten; 3–5 % der Fälle zeigen eine Progression in ein diffuses großzelliges B-Zell-Lymphom. Selten werden Milz und Knochenmark infiltriert.
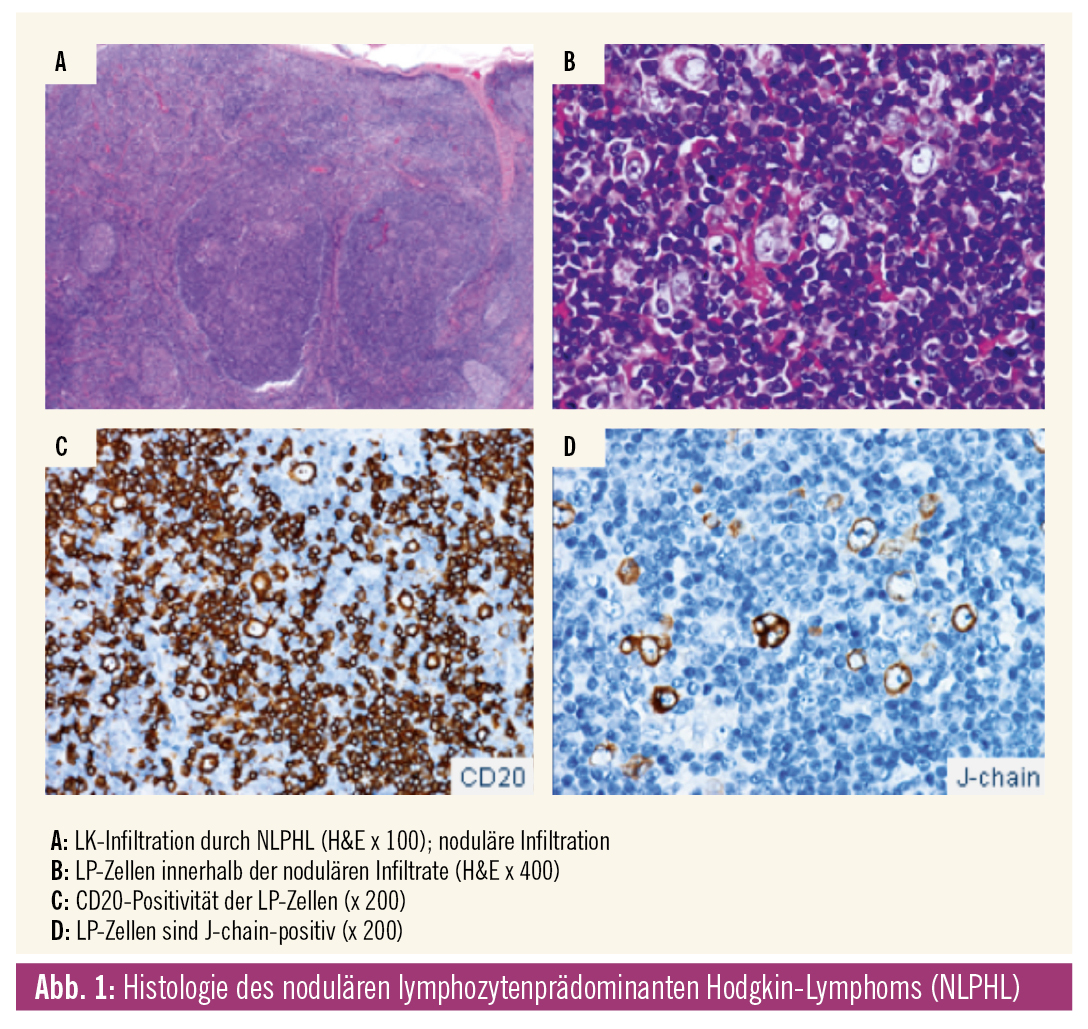
Histologisch (Abb. 1) ist die LK-Architektur partiell oder komplett aufgehoben – durch ein zumeist noduläres Infiltrat mit unregelmäßigen Netzwerken follikulärer dendritischer Retikulumzellen. Innerhalb der Noduli finden sich kleine Lymphozyten, Histiozyten und Epitheloidzellen mit untermischten Tumorzellen, genannt lymphozytenprädominante (LP) Zellen. Diese LP-Zellen sind reaktiv mit Antikörpern (Ak) gegen CD20, OCT2, CD79a, BOB1, PAX5, BCL6, CD45 sowie häufig EMA und J-chain. Die LP-Zellen sind nicht reaktiv mit Ak gegen CD15 und CD30. Wenn CD30-positive Blasten vorhanden sind, entsprechen diese reaktiven Immunoblasten. EBV-positive Tumorzellen sind nicht nachweisbar. Differenzialdiagnostisch müssen vor allem eine reaktive Progression von Keimzentren (PTKZ) und das aggressive T-Zell-histiozytenreiche großzellige B-Zell-Lymphom (TCHRBCL) abgegrenzt werden.
cHL: Die Ursprungszelle ist eine Keimzentrums-B-Zelle. Das Hodgkin-Lymphom zeigt einen großen Häufigkeitsgipfel zwischen dem 20. und 30. Lebensjahr und einen kleineren Gipfel nach dem 65. Lebensjahr. Diese ungewöhnliche Altersverteilung macht das insgesamt seltene Hodgkin-Lymphom zu einer der häufigsten hämatologischen Neoplasien bei jungen Erwachsenen.
Histologisch werden 4 Subtypen unterschieden, wobei der häufigste der nodulär sklerosierende Subtyp (Abb. 2) ist. Dieser macht etwa 70 % aller cHL in den westlichen Ländern aus. Häufig ist ein mediastinaler Befall (80 %), das Knochenmark ist bei etwa 3–5 % der Patienten infiltriert. Befallene LK zeigen ein knotiges/noduläres Infiltrat, welches von Kollagenfasern (Sklerose) umgeben ist; auch die LK-Kapsel ist meist verdickt. Die HRS-Zellen sind eingebettet zwischen reichlich reaktiven Lymphozyten, Plasmazellen, Eosinophilen und histiozytären Zellen. Durch Formalinfixierung zeigen HRS-Zellen oftmals artifiziell ein optisch leeres Zytoplasma – diese Tumorzellen werden Lakunarzellen genannt. Die HRS-Zellen sind in allen Subtypen der cHL positiv mit Ak gegen CD30 und MUM1 sowie PD1-L1. Meist besteht eine Positivität mit Ak gegen CD15 und PAX5; oft findet man eine Positivität mit B-Zell-Markern, vor allem CD20 und CD23. EBV-positive Tumorzellen werden bei den verschiedenen Subtypen in variabler Anzahl gefunden.
Die weiteren Subtypen des cHL sind: der lymphozytenreiche Subtyp, der Mischtyp und der extremst seltene lymphozytenarme Subtyp. Der lymphozytenreiche Subtyp zeigt meist ein noduläres Wachstumsmuster mit häufig exzentrisch gelegenen Keimzentren. HRS-Zellen liegen meist innerhalb dieser Noduli. Der Mischtyp weist eine aufgehobene LK-Architektur auf und es finden sich zarte Fibrosebänder. Das inflammatorische Begleitinfiltrat zeigt eine sehr variable Zusammensetzung, oftmals mit reichlich untermischten Epitheloidzellen. Der extrem seltene, oft mit HIV-Infektion kombinierte lymphozytenarme Subtyp muss von einem ALK-negativen anaplastischen großzelligen Lymphom unterschieden werden.

Resümee
Das Hodgkin-Lymphom besteht aus 2 Gruppen: dem NLPHL und dem cHL mit seinen 4 Subtypen. Die Tumorzellen des NLPHL, die LP-Zellen, sind positiv mit Ak gegen CD20, OCT2, CD79a, BOB1, PAX5, BCL6 und CD45 sowie häufig EMA und J-chain. CD30 ist negativ und EBV-positive Zellen sind so gut wie nie nachweisbar. Die hauptsächlichen Differenzialdiagnosen sind das aggressive TCHRBCL und die reaktive PTKZ.
Die Tumorzellen des cHL sind immer positiv mit Ak gegen CD30, MUM1 und PD1-L1, meist positiv mit Ak gegen CD15 und in etwa 20 % der Fälle positiv mit Ak gegen CD20 und CD23. EBV-positive Tumorzellen können in allen Subtypen in variabler Anzahl vorkommen. Differenzialdiagnostisch sind vor allem EBV-assoziierte Erkrankungen, insbesondere das EBV-positive mukokutane Ulkus, abzugrenzen.
1 Swerdlow SH et al., World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissues; Lyon 2017

AutorIn: ao. Univ.-Prof. Dr. Christine Beham-Schmid
Institut für Pathologie, Medizinische Universität Graz

Ursprünglich erschienen:
Patho 01|2018
Patho 01|2018