


























































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Alzheimer-Demenz: Update aus der pharmakologischen Forschung
31. Oktober 2012

Die Alzheimer-Demenz (AD) ist die häufigste neurodegenerative Erkrankung und nimmt mit ca. 50 % den größten Anteil unter den Demenzen ein. Weltweit sind mehr als 35 Millionen Menschen von dieser Erkrankung betroffen, und die Anzahl wird in den nächsten Jahren aufgrund der steigenden Lebenserwartung noch weiter zunehmen.
Bis dato stehen zur Behandlung der AD mit den Azetylcholinesterasehemmern (AChE-I) und Memantin mehrere gut verträgliche, jedoch nur symptomatisch wirkende medikamentöse Therapien zur Verfügung. Neue innovative Therapieoptionen, die auf die Pathophysiologie der AD abzielen und krankheitsmodifizierend wirken sollen, sind derzeit Gegenstand intensiver Forschung.
Vielfach wird nun angenommen, dass die AD multifaktoriell bedingt ist, beziehungsweise das klinische Bild und die neuropathologischen Befunde bei einer AD die gemeinsame letzte Endstrecke unterschiedlicher Ätiologien darstellen. So wurde eine Vielzahl molekularer Läsionen entdeckt, wobei im Wesentlichen fehlgefaltete Proteine im alternden Gehirn oxidative und inflammatorische Schäden verursachen dürften, welche zu synaptischen Dysfunktionen, Veränderungen im Energiehaushalt und zur Apoptose der Neurone führen1. Im Folgenden werden die unterschiedlichen pathophysiologischen Theorien mit den daraus resultierenden Therapieansätzen zusammengefasst und aktuell beforschte Substanzen dargestellt2.
β-Amyloid
Bei der AD findet man zerebrale Plaques, die aus Amyloid-β-Peptiden (Aβ) bestehen. Im sogenannten amyloidogenen Weg wird Aβ, physiologisch vorkommende Peptide, die eine wichtige Funktion bei der Informationsverarbeitung im Gehirn haben, von proteinspaltenden Sekretasen (β- und γ-Sekretasen) aus dem Amyloid-Vorläuferprotein (APP), einem integralen Membranprotein, abgebaut. Hierbei entstehen verschieden lange Aβ-Peptide: Das Aβ40, welches 40 Aminosäuren lang ist, und zu einem kleineren Teil Aβ42 mit 42 Aminosäuren, welches aber stärker zur Aggregation zu Oligomeren neigt und sich damit in Form von Plaques ablagert. Ein weiterer Teil von APP wird im nicht amyloidogenen Weg von der α-Sekretase an anderen Stellen im Protein zu Fragmenten abgebaut, die keine Aggregationstendenzen zeigen.
Aβ wirkt toxisch auf Neurone und kann deren Apoptose auslösen. Weiters begünstigt Aβ die im Alter vor allem im Hippocampus auftretende, natürliche Reduktion von Synapsen und die damit zusammenhängende Beeinträchtigung der Impulsübertragung. Hierzu ergeben sich drei Therapiebereiche: die Förderung des Aβ-Abbaus, die Reduktion der Aβ-Produktion und die Hemmung der Aβ-Aggregation.
Förderung des Aβ-Abbaus
Aktuell werden diesbezüglich immunologische Therapieansätze intensiv beforscht. Bei der aktiven Immunisierung durch Impfung wird das Immunsystem mittels einer Typ-2-Immunantwort zur Bildung von Antikörpern gegen Aβ-Epitope angeregt, damit soll der Abbau von Aβ-Plaques gefördert werden. Bei der passiven Immunisierung werden Antikörper gegen Aβ intravenös verabreicht.
Die Entwicklung der ersten aktiven Immunisierung gegen β-Amyloid, musste zunächst im Jahr 2002 gestoppt werden, da bei 6 % der Patienten in einer Phase-II-Studie eine aseptische Meningoenzephalitis auftrat, die auf einen Shift von einer Typ-2-Immunreaktion (Produktion von Antikörpern) zu einer proinflammatorischen Typ-1-Immunantwort (zytotoxische T-Zell-Response) zurückzuführen war. Danach wurden alternative Impfstoffe entwickelt, die eine wesentlich geringere Toxizität aufweisen. Auch die passive Immunisierung hat gelegentlich schwere Nebenwirkungen, wie zerebrale vasogene Ödeme und Mikrohämorrhagien, zur Folge.
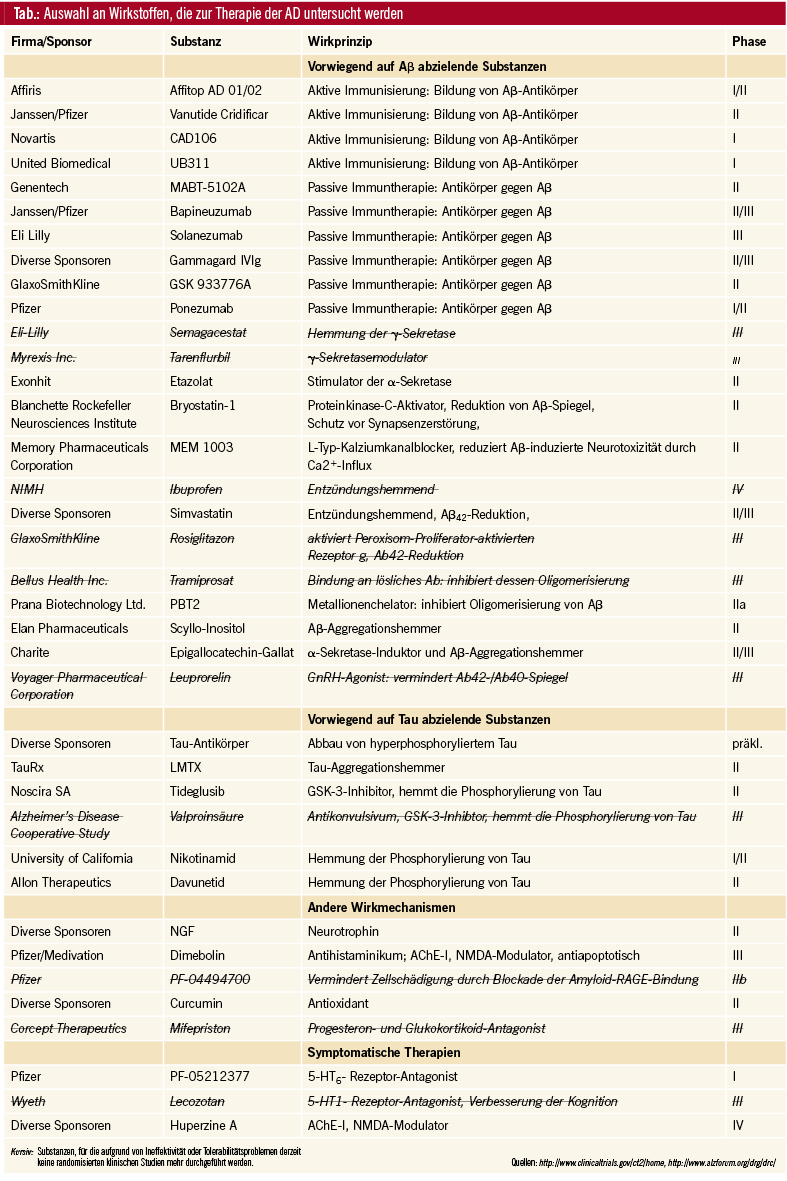
In Zusammenhang mit der aktiven als auch der passiven Immunisierung werden von mehreren Firmen zahlreiche Wirkstoffe untersucht, die sich bereits teilweise in Phase III befinden (Tab.). Die bis dato vorliegenden Ergebnisse zur aktiven und passiven Immunisierung zeigen Großteils übereinstimmend eine Reduktion der Aβ-Menge im Gehirn bei AD-Patienten, was jedoch bei bereits bestehender AD, im Gegensatz zu Ergebnissen im Tiermodell, üblicherweise nicht mit einer kognitiven Verbesserung einhergeht. Es wird spekuliert, dass eine Wirksamkeit in noch nicht klinisch auffälligen bzw. sehr frühen Stadien der AD vorhanden sein könnte.
Eine rezente präklinische Studie mit Bexaroten, das bereits zur Behandlung des kutanen T-Zell-Lymphoms zugelassen ist, zeigt, dass sich bei transgenen „Alzheimer-Mäusen“, die mit diesem Wirkstoff behandelt wurden, nicht nur das Ausmaß der Aβ-Plaques bereits innerhalb von 72 Stunden um 50 % und nach 14 Tagen um 75 % reduzierte, sondern sich auch kognitive Symptome und Verhaltensstörungen besserten. Bexaroten ist ein Agonist am Retinoid-X-Rezeptor und beeinflusst so die Expression von Apolipoprotein E, das somit in größeren Mengen produziert wird und den Abbau von Aβ-Plaques begünstigt.
Reduktion der Aβ-Produktion
Eine weitere Aβ-beeinflussende Wirkstoffgruppe sind γ-Sekretasehemmer (z. B. Semagacestat) und der γ-Sekretasemodulator Tarenflurbil, die jedoch in klinischen Studien mehr als enttäuschende Ergebnisse zeigten. So verschlechterten sich die kognitiven Funktionen in der Verumgruppe einer Phase-III-Studie mit Semagacestat sogar, und die Einnahme desselbigen erhöhte auch das Risiko für maligne Hauterkrankungen, sodass die weitere Entwicklung für alle γ-Sekretasehemmer und modulatoren gestoppt bzw. pausiert wurde.
Eine alternative Möglichkeit, die Aβ-Produktion zu minimieren, ist eine Stimulierung der α-Sekretase. Etazolat, ein selektiver GABAA-Rezeptor-Modulator, der die α-Sekretase stimuliert, wird in Phase-II-Studien getestet. Epigallocatechin-Gallat (EGCg) ist ein Polyphenol aus dem grünen Tee, ebenfalls ein Induktor der α-Sekretase, und fördert somit den Abbau von APP im nichtamyloidogenen Weg. Zusätzlich hemmt es die Akkumulation von Aβ. Phase-II- und -III-Studien zur Wirksamkeit bei AD-Patienten laufen.
Insulin-Sensitizer: Diabetes mellitus Typ 2 (DM) scheint ebenfalls ein Risikofaktor für die Entstehung einer AD sein. Ein defekter Insulinstoffwechsel erzeugt einen neuronalen Energiemangel und macht diesen anfällig für oxidative Schäden und beeinträchtigt die Neuroplastizität. In diesem Zusammenhang wurden die bereits zur Behandlung des DM zugelassene Insulin-Sensitizer Rosiglitazon und Pioglitazon, die als β-Sekretasehemmer wirken, indem sie im Zellkern den PPAR-Rezeptor („peroxisome proliferator-activated receptor“) vom Typ γ aktivieren, untersucht.
Die Aktivierung des PPARγ-Rezeptors erhöht die Insulin-Empfindlichkeit der Zellen von Leber, Muskulatur und Fettgewebe, was somit zu einer Senkung der Insulinresistenz und über die Hemmung der β-Sekretase im Tiermodell zu reduzierten Aβ42-Spiegeln führt. In zwei kleinen Studien hat sich gezeigt, dass Rosiglitazon bei Non-ApoE-ε4-Allelträgern, einem Polymorphismus des ApoE-ε4-Gens, der mit einer niederen Inzidenz an frühzeitig auftretenden AD einhergeht, geringe aber signifikante Verbesserungen der Kognition bewirkte, aber bei ApoE-ε4-Allelträgern keinen Effekt bzw sogar zu einer Verschlechterung der Kognition führte. Aufgrund der nicht überzeugenden Ergebnisse und der potenziell schweren kardiovaskulären Nebenwirkungen wurde die weitere Untersuchung der Insulin-Sensitizer zur Behandlung der AD eingestellt.
Isproniclin, Bryostatin-1: Aβ ist zusammen mit Tau im Rahmen der AD zudem mit dem Mangel der cholinergen Signalübertragung vergesellschaftet. Hierbei bindet Aβ an die präsynaptischen α-7-nikotinischen Azetylcholinrezeptoren und behindert somit die Ausschüttung von Azetylcholin, was in weiterer Folge die für die Gedächtnisfunktionen relevante „Langzeit-Potenzierung“ beeinträchtigt. Agonisten an nikotinischen Rezeptoren, wie Isproniclin werden derzeit hinsichtlich einer Verbesserung der Kognition in Phase II untersucht.
Pharmakologische Stimulierung am postsynaptischen muskarinischen Rezeptor 1 (M1) aktiviert die Proteinkinase C, was die Weiterverarbeitung von APP im nicht amyloidogenen Weg fördert und die Phosphorylierung von Tau reduziert. Klinische Studien mit selektiven M1-Agonisten (z. B. Cevimelin, LU25-109, NGX267), die teilweise auch zur Therapie der Xerostomie angewendet werden, zeigten eine Verbesserung der Kognition und eine Reduktion der Aβ-Spiegel im Liquor. Diese Wirkstoffe werden jedoch aufgrund von intolerablen Nebenwirkungen nicht weiter beforscht.
Bryostatin-1 ist ebenfalls ein Proteinkinase-C-Aktivator und reduzierte im Mausmodell Aβ-Spiegel, war präventiv hinsichtlich des damit verbundenen Verlustes von Synapsen und schützte vor kognitivem Abbau. Bryostatin-1 befindet sich derzeit in der klinischen Phase II.
Kalziumstoffwechsel: Bei der AD findet man weiters eine Störung des Kalziumstoffwechsels im Gehirn. Neuronale Kalziumspiegel werden durch L-Typ-Kalziumkanäle reguliert. Eine anormale Funktion dieser Kanäle ist mit einer Beeinträchtigung der für Gedächtnis und andere kognitive Funktionen zuständigen Bahnen verantwortlich und kann eventuell sogar einen neuronalen Zelltod verursachen. Nimodipin, ein selektiver L-Typ-Kalziumkanalblocker, schützte im Tiermodell mit Ratten die kortikalen Neurone vor Aβ-induziertem Ca2+-Influx, hemmte die DNA-Fragmentation, was ein für die Apoptose typisches Charakteristikum ist, und reduzierte den Zelltod. Nimodipin zeigte darüber hinaus in einigen Studien Wirksamkeit hinsichtlich Verbesserung kognitiver Funktionen und reduzierte Aβ42-Spiegel in Rattenneuronen. Da Nimodipin aufgrund von Nebenwirkungen (Blutdrucksenkung) nur vorsichtig dosiert werden kann, wird aktuell eine Nachfolgersubstanz mit gleicher Pharmakodynamik, nämlich MEM 1003, entwickelt, das in Phase-I- und -II-Studien keinen Effekt auf den Blutdruck hatte.
Statine, NSAR: Zahlreiche Untersuchungen im Tiermodell und in vitro haben demonstriert, dass die Senkung der Cholesterinkonzentration durch Statine zu einer verminderten Freisetzung von Aβ führt und die Neuroinflammation zu reduzieren vermag. In einzelnen epidemiologischen Studien wurde beobachtet, dass in Patientengruppen, die mit Statinen behandelt wurden, das Neuauftreten einer AD um 60–70 % erniedrigt war. Es wurde weiters gezeigt, dass Simvastatin die Konzentration von Aβ im Liquor bei Patienten mit milden Formen der AD geringgradig verringerte. Mehrere Studien zur Wirksamkeit bei AD-Patienten konnten keine überzeugenden Ergebnisse präsentieren, sodass der Einsatz von Statinen bei der AD derzeit nicht empfohlen wird. Weitere Studien sind noch abzuwarten.
Dasselbe gilt für NSAR wie Ibuprofen, wo sich in retrospektiven Studien zur Langzeiteinnahme Hinweise auf eine Reduktion beziehungsweise eine Verzögerung eines AD-Krankheitsausbruchs ergaben. Zudem wurde noch Phenserin, ein AChE-I, der auch die Translation der mRNA von APP und in weiter Folge Aβ-Spiegel senkt, in klinischen Studien geprüft. Phenserin war gut verträglich, wird jedoch wegen fehlenden positiven Ergebnissen nicht mehr geprüft.
LH-Sekretion: Da Frauen häufiger von einer AD betroffen sind, kann man vermuten, dass auch hormonelle Faktoren in der AD-Pathogenese eine Rolle spielen. Die Plasmaspiegel des luteinisierenden Hormons sind bei Frauen signifikant höher als bei Männern. Der Hippokampus, der bei der AD unter anderem am meisten betroffen ist, weist eine hohe Dichte an LH-Rezeptoren auf. In Hinblick auf die Wirksamkeit bei der AD wurde Leuprorelin, ein GnRH-Analogon, das zu einer Absenkung der LH-Sekretion führt, bereits eingehend untersucht. Im Mausmodell verminderte Leuprorelin die Aβ-Spiegel, was mit einer kognitiven Verbesserung korrelierte. Wegen nichtsdestotrotz unzureichender Wirkung in klinischen Phase-II- und -III-Studien werden keine randomisierten klinischen Studien zu Leuprorelin mehr durchgeführt.
Hemmung der Aβ-Aggregation
Andere Substanzen hemmen durch unterschiedliche Mechanismen die Oligomerisation von Aβ. Diesbezüglich zu nennen ist unter anderem Tramiprosat, das an lösliches Aβ bindet und somit sowohl der Bildung als auch in weiterer Folge der Ablagerung von Aβ-Plaques im Gehirn vorbeugt. Für Tramiprosat wurde auch die Prävention der zerebralen Amyloidangiopathie spekuliert. Klinische Studien bis Phase-III zeigten jedoch unzureichende Wirksamkeit, sodass die weitere Entwicklung eingestellt wurde.
Ein weiteres Wirkprinzip stellen Metallionenchelatoren, wie Clioquinol (PBT1) und die Weiterentwicklung PBT2 dar, die die Bindung der Metallkationen Zink und Kupfer an Aβ und die Aggregation von Aβ-Plaques hemmen. In einer Phase-IIa-Studie mit PBT2 konnte eine dosisabhängige Reduktion der Aβ-Spiegel im Liquor und eine Verbesserung der exekutiven Funktionen gezeigt werden.
Scyllo-Inositol ist ein vor allem in der Kokospalme vorkommender Zuckeralkohol, der im Mausmodell die Entstehung von Aβ-Plaques verhindert und Gedächtnisstörungen zu verbessern vermag. Aktuell befindet sich die Entwicklung von Scyllo-Inositol in der klinischen Phase II.
Tau
Neben Aβ findet man bei der AD andererseits auch intrazellulär gelegene Neurofibrillen, die aus hyperphosphorilisiertem Tau-Protein bestehen. Tau-Proteine sind mikrotubuliassoziierte Proteine, die eine wichtige Rolle in der Bildung des Zytoskeletts spielen. Mutationen im MAPT-Gen, das für das Tau-Protein kodiert, können beim Menschen eine Vielzahl von erblichen Erkrankungen wie die AD, die Pick-Krankheit, die kortikobasale Degeneration oder die progressive supranukleäre Blickparese verursachen, wobei jeweils verschiedene Typen von Neurofibrillenbündeln gefunden werden.
Neurodegenerative Erkrankungen mit Ablagerungen von Tau-Protein werden in der Gruppe der Tauopathien zusammengefasst. Beim Zusammenbau in filamentäre Strukturen werden die einzelnen Tau-Proteine über ihre sich wiederholenden Abschnitte miteinander verknüpft, wobei das aminoterminale Ende abgeschnitten wird. Eine Hyperphoshorylierung der gepaarten helikalen Tau-Filamente führt zu einem funktionsunfähigen Protein, das Mikrotubuli nicht mehr stabilisieren kann, und somit zu einer Störung des intrazellulären und intraaxonalen Transportes. Intermittierende Aggregate aus abnormalen Tau-Molekülen entfalten zytotoxische Effekte und sind mit kognitiven Dysfunktionen vergesellschaftet.
Hier kommen drei Therapieansatzpunkte in Betracht: Förderung des Abbaus von Tau-Aggregaten, Tau-Aggregationshemmer und Hemmung der Tau-Hyperphosphorylierung.
Förderung des Abbaus von Tau-Aggregaten
Derzeit befinden sich auch immunologische Therapieansätze für die Beseitigung von hyperphosphorylierten Tau-Proteinaggregaten in der präklinischen Entwicklungsphase. Im Maus-Modell konnte gezeigt werden, dass Anti-Tau-Antikörper pathologische Tau-Aggregate reduzierten und dies zu einer deutlichen Verbesserung der kognitiven Funktionen führte. Die Antikörper werden über Pinozytose (rezeptorvermittelte oder Fluid-Phase-Endozytose) in die Neurone aufgenommen und binden selektiv an hyperphosphoryliertes Tau. Es wird spekuliert, dass die antikörpermarkierten Tau-Proteine lysosomal abgebaut werden. Normales Tau-Protein bleibt unbeeinflusst. Nach bisherigem Wissensstand sind keine zytotoxischen Effekte zu erwarten.
Tau-Aggregationshemmer
In dieser Indikation wurde der Einsatz von Methylthioninium (Methylenblau) untersucht. Es wirkt als Tau-Aggregationshemmer. Darüber hinaus gibt es Hinweise, dass Methylthioninium den Abbau abnormer Tau-Proteine mittels Autophagozytose induziert. Weiters wurde berichtet, dass durch Methylthioninium im Mausmodell auch die Aβ-Spiegel reduziert wurden. Es verzögert die Krankheitsprogression und verbessert die kognitiven Funktionen nach 6-monatiger Einnahme, wie bereits in einer Phase-IIb-Studie gezeigt werden konnte. Aufgrund der schlechten Verträglichkeit von Methylthioninium liegt der Fokus nun in der Untersuchung von Tau-Aggregationshemmern der zweiten Generation mit ähnlicher Molekularstruktur und gleichem Wirkmechanismus, wie LMTX.
Hemmung der Tau-Hyperphosphorylierung
Die Glykogen-Synthase-Kinase-3 (GSK-3) ist verantwortlich für die Phosphorylierung von Tau-Proteinen. Für das Antikonvulsivum Valproinsäure und für Lithium wurde eine neuroprotektive Wirkung in Zusammenhang mit einer GSK-3-Hemmung spekuliert. Alleine Valproinsäure schaffte es bisher in Phase III, jedoch mit eher enttäuschenden Ergebnissen. Darüber hinaus befindet sich mit Tideglusib ein weiterer GSK-3-Inhibitor in der klinischen Überprüfung (Phase II). Tideglusib wird auch zur Therapie der supranukleären progressiven Paralyse getestet.
Für Nikotinamid (Vitamin B3) konnte auch eine Reduktion der Konzentration von hyperphosphoryliertem Tau in Mäusegehirnen nachgewiesen werden. In einigen Studien wurde eine gute Verträglichkeit gezeigt, und derzeit laufen klinische Studien zur Wirksamkeit bei der AD. Auch für Davunetid, das intranasal verabreicht wird, wurde eine Hemmung der Tau-Hyperphophorylierung sowie ein Schutz der Neuronen vor Aβ-Toxizität berichtet. In Phase-II-Studien konnte eine gute Verträglichkeit und eine leichte Verbesserung der kognitiven Funktionen bei Patienten mit Mild Cognitive Impairment erfasst werden.
Andere Mechanismen
Neurotrophine
Neurogenese kann auch im erwachsenen Gehirn, beispielsweise als Antwort auf eine Schädigung, stattfinden. Das Wachstum und Überleben der cholinergen Neurone des basalen Vorderhirns sind vom „nerve growth factor“ (NGF) abhängig, und es wurden Zusammenhänge einer NGF-Dysbalance mit der Aktivierung der amyloidogenen Kaskade berichtet. Eine zielgerichtete Behandlung des Vorderhirns von Tieren mit NGF führte zu Hemmung des neuronalen Todes, stimulierte die synaptischen cholinergen Funktionen und kognitive Verbesserungen.
Bei der Behandlung mit NGF stellt der Transport an den Wirkort die Herausforderung dar. In den ersten Studien an AD-Patienten erfolgte eine intrazerebroventrikuläre Infusion, gefolgt von autologen intrazerebralen Infusionen mit gentechnisch veränderten NGF-produzierenden Fibroblasten und intrazerebralen stereotaktischen Implantationen von Kathetern, die NGF-produzierende Zellen enthalten. In einer Studie an 6 AD-Patienten mit einem derartigen Implantat, das sie für 12 Monate erhielten, zeigte sich bei guter Sicherheit und Verträglichkeit eine Vermehrung der kortikalen nikotinischen Rezeptoren und eine kognitive Verbesserung. Intranasale und topische Applikationsformen auf die Augenoberfläche sind derzeit in der präklinischen Entwicklungsphase.
Apoptosehemmung
Es hat sich gezeigt, dass Aβ toxisch auf Mitochondrien wirkt. In Gehirnen von AD-Patienten und transgenen Mäusen fanden sich Akkumulationen von Aβ in zerstörten neuronalen Mitochondrien. In weiterer Folge wird die Zytochrom-C-Oxidase in Mitleidenschaft gezogen, woraufhin der Elektronentransport, die ATP-Produktion, der Sauerstoffaustausch und das mitochondriale Membranpotenzial gestört werden, was im Endeffekt wieder zur neuronalen Apoptose führt.
Hier setzt das Antihistaminikum Dimebolin (Latrepirdin) an, das ein potenzieller Mitochondrienstimulator ist und dadurch die mitochondrial vermittelte Apoptosehemmung fördert. In Studien haben sich darüber hinaus cholinesterasehemmende Effekte und eine antagonistische Wirkung am NMDA-Rezeptor für diesen Wirkstoff gezeigt. Aktuell befindet sich Dimebolin in Phase III, nachdem in Phase-II-Studien die Wirksamkeit in Hinsicht auf kognitive Verbesserung vermutet wurde.
Der RAGE-Inhibitor (PF-04494700) hemmt die Bindung von Aβ am „receptor for advanced glycation endpoints“ (RAGE) und schützt somit vor toxischen Effekten an Neuronen, der Mikroglia und der Blut-Hirn-Schranke. Die Ergebnisse von mehreren Phase-II-Studien waren nicht überzeugend, sodass Pfizer die Entwicklung einstellte.
Antioxidantien
Eine antidementive Wirkweise hat man auch bei Antioxidantien (Vitamin E, Curcumin, Östrogen) angedacht, diese erwiesen sich jedoch bisher generell als gering bis gar nicht wirksam. Derzeit laufen noch diverse Phase-II- und -III-Studien mit Curcumin und Vitamin E in Kombination mit anderen Nahrungsergänzungsmitteln bei Mild Cognitive Impairment und AD.
Mifepriston (RU486), ein Progesteron- und Glukokortikoid-Antagonist wurde als „Abtreibungspille“ bekannt. Da Mifepriston in der Lage ist, die Wirkung von Kortisol zu antagonisieren und Kortisol an der Initiierung der AD beteiligt zu sein scheint, sind neuroprotektive und antioxidative Effekte zu vermuten. In einer placebokontrollierten Studie zeigten die AD-Patienten aus der Verumgruppe eine Verbesserung der kognitiven Funktionen. Größere Studien werden derzeit nicht durchgeführt.
Antikonvulsiva
Die AD ist mit einer erhöhten Inzidenz von epileptischen Anfällen vergesellschaftet. Palop und Mucke hypothetisieren mithilfe von Beobachtungen im Mausmodell, dass erhöhte Aβ-Spiegel zu einer abnormen exzitatorischen neuronalen Aktivität führen, welche in weiterer Folge eine kompensatorische inhibitorische Antwort, die Lernen und Gedächtnis beinhaltet, verursacht. Beide Veränderungen führen zu einem kognitiven Abbau. Bisher wurden klinische Studien mit Antikonvulsiva wie Valproinsäure in ihrer Eigenschaft als GSK-3-Inhibitor (s. o.) und zur Behandlung von Agitation bei der AD in späteren klinischen Stadien durchgeführt, die keine positiven Ergebnisse hinsichtlich der kognitiven Symptomatik zeigten. Untersuchungen in frühen Stadien der AD fehlen noch.
Neue symptomatische Therapieansätze
Derzeit befinden sich 5-HT6-Rezeptor-Antagonisten (PF-05212377, SAM-760, SB-742457) in Phase I. Im Tiermodell konnte gezeigt werden, dass eine Blockade am 5-HT6-Rezeptor, die in weiterer Folge die cholinerge, glutamaterge, noradrenerge und dopaminerge Neurotransmission beeinflusst, eine Verbesserung der kognitiven Funktionen bewirkte und zudem auch eine anxiolytische und antidepressive Wirkung entfaltet.
Auch über das serotonerge System wirken die selektiven 5-HT1A-Rezeptor-Antagonisten Lecozotan und Xaliproden, die die Glutamat- und Azetylcholinfreisetzung im Hippokampus potenzieren. Bei gealterten Rhesusaffen verbesserte Lecozotan die kognitiven Funktionen und konnte pharmakologisch induzierte Lerndefizite rückgängig machen. In klinischen Studien konnte jedoch keine ausreichende Wirksamkeit für beide Wirkstoffe nachgewiesen werden, sodass diese Wirkweise nicht weiterverfolgt wird.
Auch verschiedene Naturheilverfahren haben in der modernen Arzneimittelforschung Interesse geweckt. Das Bärlappgewächs Huperzia serrata wird in der chinesischen Medizin eingesetzt. Aus dieser Pflanze wird der Wirkstoff Huperzine A gewonnen. In Studien konnte gezeigt werden, dass die Substanz als AChE-I- und NMDA-Rezeptorantagonist fungiert, aber auch neuroprotektiv und antiinflammatorisch wirkt.
Resümee
Neue innovative Therapieoptionen, die auf die Pathophysiologie der AD abzielen, sind derzeit Gegenstand intensiver Forschung. Nur wenige dieser neuen Substanzen werden den Weg bis zur Zulassung schaffen, da bei einem Großteil der Substanzen die weitere Entwicklung aufgrund von Ineffektivität oder Toxizität bzw. Tolerabilitätsproblemen aufgegeben werden muss bzw. schon aufgegeben werden musste. Die noch immer fehlende Kenntnis der genauen Pathophysiologie der AD erschwert alle Therapiebemühungen.
1 Querfurth HW, LaFerla FM, Alzheimer’s disease. N Engl J Med 2010; 362(4):329–44.
2 Mangialasche F et al., Alzheimer’s disease: clinical trials and drug development. Lancet Neurol 2010; 9(7):702–16.
Weitere Literatur bei den Verfassern

Ursprünglich erschienen:
SP 03|2012
SP 03|2012