


























































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Selten, aber häufig heilbar – Hodgkin-Lymphome
20. August 2011
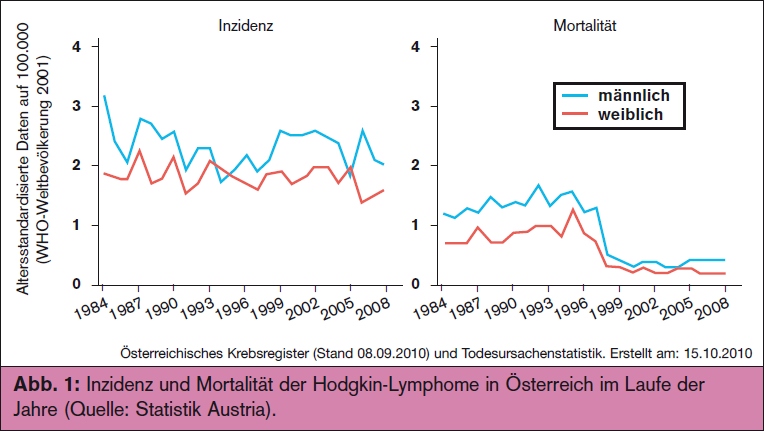
Mit einer Inzidenz von 2-3 Fällen auf 100.000 Personen machen Hodgkin-Lymphome etwa 10% aller Lymphomerkrankungen aus. In Österreich kann somit jährlich mit ca. 160 Neuerkrankungen und ungefähr 35 Todesfällen gerechnet werden. Das mediane Alter bei Erstdiagnose beträgt 32 Jahre mit einer bimodalen Altersverteilungskurve mit einem Gipfel bei ca. 25 sowie bei ca. 65 Jahren. Die Inzidenz ist in den Industrieländern in den letzten Jahren tendenziell rückläufig (> Abb. 1).
Klinisches Bild
Etwa 70% der Patienten präsentieren sich bei Erstdiagnose mit einer palpablen pathologischen Lymphadenopathie, wobei die Zervikal-/ Supraklavikularregion am häufigsten betroffen ist (∼60-80%), gefolgt von axillärem (∼10-20%) und inguinalem (∼5-10%) Befall. Mediastinale und retroperitoneale Regionen sind in ∼60-70% bzw. ∼25% der Fälle mitbetroffen. Die Mehrzahl der Patienten weist außerdem systemische Krankheitszeichen wie Leistungsknick, B-Symptomatik (Fieber, Gewichtsverlust, Nachtschweiß) oder Pruritus auf. Der in manchen Lehrbüchern immer noch als typisch beschriebene “Alkoholschmerz” der betroffenen Lymphknoten tritt hingegen nur selten auf (< 10%). In ca. 2-5% der Fälle werden auch extralymphatische Organe (z. B. Leber, Niere, Haut) infiltriert, was entsprechende organtypische Symptome hervorrufen kann. Noch seltener (1%) kommt es zum Auftreten von paraneoplastischen Syndromen wie Hyperkalzämie oder neurologischen Symptomen.
Diagnose und Klassifikation
Charakteristischerweise finden sich beim klassischen Hodgkin-Lymphom nur wenige Hodgkin-Reed-Sternberg-Zellen auf einem zellreichen reaktiven Hintergrund. Um dem Pathologen ausreichend Material zur Verfügung zu stellen, sollte eine Lymphknoten-Exzision oder alternativ eine Stanzbiopsie mit einer großlumigen Nadel durchgeführt werden. Von einer Feinnadelbiopsie wird abgeraten. Da die Diagnosestellung für den Pathologen sehr
schwierig sein kann, sollte eine Beurteilung durch einen erfahrenen Referenzpathologen angestrebt werden. Nach der aktuellen WHO-Klassifikation werden folgende Formen der Hodgkin-Lymphome unterschieden:
- klassisches Hodgkin-Lymphom mit den Untergruppen
- nodulär sklerosierender Subtyp (∼70% der Fälle)
- Mischtyp (∼20-25%)
- lymphozytenreicher klassischer Subtyp (∼5%)
- lymphozytenarmer Subtyp (< 1%)
- lymphozytenprädominantes Hodgkin-Lymphom (synonym: noduläres Paragranulom); ca. 5% aller Hodgkin-Lymphome
Stadieneinteilung und Risikogruppen
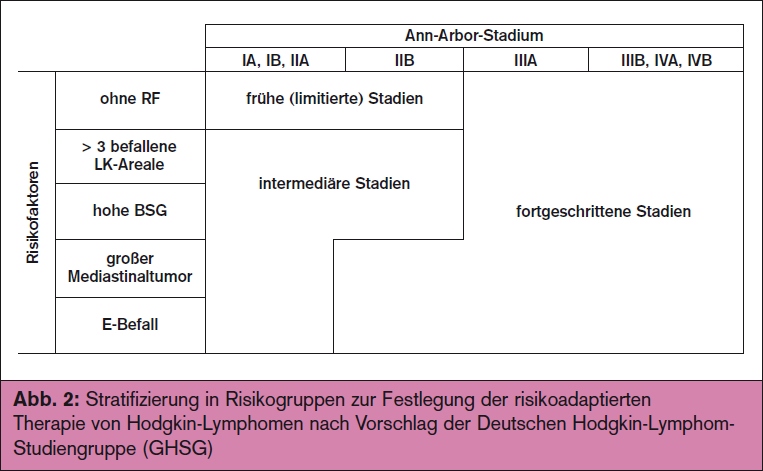
Die Stadieneinteilung erfolgt nach einem modifizierten Ann-Arbor-Schema (> Tab.) Zusätzlich wird gemäß der deutschen Hodgkin-Studiengruppe (GHSG) eine Stratifizierung nach folgenden prognostisch relevanten Risikofaktoren vorgenommen:
- Befall von ≥ 3 Lymphknotenarealen
- BSG-Erhöhung (≥ 30 mm/h ohne B-Symptome; ≥ 50 mm/h bei B-Symptomatik)
- großer Mediastinaltumor (≥ 1/3 des Thoraxdurchmessers im Röntgen)
- E-Befall
Diese Kriterien werden in ähnlicher Form auch von anderen Studiengruppen verwendet. Unter Berücksichtigung von Stadium und Risikofaktoren erfolgt eine therapierelevante Einteilung in frühe, intermediäre und fortgeschrittene Stadien (> Abb. 2).

Risikoadaptierte Therapie
Obwohl Hodgkin-Lymphome mittlerweile zu den am besten heilbaren Malignomen überhaupt gehören, sollte zur weiteren Therapieoptimierung im Sinne einer Reduktion von Langzeittoxizitäten (Infertilität, Sekundärmalignome, Herz- oder Lungenerkrankungen) wann immer möglich eine Therapie im Rahmen von klinischen Studien angestrebt werden. In
Österreich beteiligen sich eine ganze Reihe von Zentren an den Studien der GHSG, die durch die Arbeitsgruppe Medikamentöse Tumortherapie (AGMT) koordiniert werden (www.agmt.at; Coordinating Investigator: Prof. Richard Greil, Salzburg). Die Therapie erfolgt streng risikoadaptiert, sodass ein sorgfältiges Staging inklusive Anamnese (B-Symptome?), Labor inkl. BSG, Thoraxröntgen, Hals-Thorax-Abdomen-CT, Knochenmarksbiopsie sowie eine präzise Einteilung in die jeweilige Risikogruppe (frühes vs. intermediäres vs. fortgeschrittenes Stadium) nach > Abb. 2 obligat ist.
Therapie für frühe Stadien: Frühe Stadien haben eine exzellente Prognose. So betrug beispielsweise in der kürzlich publizierten HD-10-Studie der GHSG das progressionsfreie Überleben 87% und das Gesamtüberleben sogar 95% (Follow-up: 8 Jahre). In dieser Studie konnte auch ein neuer Therapiestandard festgelegt werden: 2 Zyklen Chemotherapie nach dem ABVD-Protokoll, gefolgt von einer so genannten Involved-Field-Radiotherapie (IF-RT, Bestrahlung aller betroffenen Lymphknotenregionen) von 20 Gy.
Therapie für intermediäre Stadien: Patienten in intermediären Stadien haben ebenfalls eine exzellente Prognose (progressionsfreies und Gesamtüberleben nach 5 Jahren 90% bzw. > 95%). Auch hier wird eine kombinierte Vorgehensweise aus Chemotherapie (z. B. 4 Zyklen ABVD) + IF-RT angewendet. Die kürzlich präsentierten Daten der GHSG-HD-14-Studie
legen jedoch eine dosisintensivierte Behandlung nach dem “2 + 2”-Schema (2 x BEACOPPeskaliert + 2 x ABVD) + 30 Gy IFRT nahe.
Therapie für fortgeschrittene Stadien: Auch bei fortgeschrittenen Stadien beträgt das Gesamtüberleben nach 10 Jahren > 80%. Die optimale Standardtherapie wird hier jedoch noch kontroversiell diskutiert. In Frage kommen nach aktueller Datenlage einerseits 8 Zyklen ABVD, andererseits 8 x BEACOPPeskaliert, wobei letzteres aufgrund der Toxizität nicht unumstritten ist. So zielen derzeit laufende Studien vor allem auf eine weitere Toxizitätsreduktion ab, indem beispielsweise PET-stratifizierte Therapieansätze verfolgt werden. Eine Strahlentherapie sollte nur bei residuellen Tumormassen nach Abschluss der Behandlung bzw. bei Vorliegen von Lymphknoten ≥ 5 cm (“bulky disease”) erfolgen.
Besondere Therapiesituationen
- Patienten mit signifikanten Komorbiditäten bzw. > 60 Jahre: Aufgrund der Toxizität des BEACOPP-Protokolls sollten diese Patienten eine stadienadaptierte Therapie mit 2 bzw. 4 bzw. 6-8 Zyklen ABVD erhalten.
- lymphozytenprädominantes Hodgkin-Lymphom: Bei lokalisierten Stadien ist aufgrund der hervorragenden Prognose eine alleinige IF-RT mit 30 Gy indiziert, bei fortgeschrittenen Stadien eine Therapie analog zu klassischen Hodgkin-Lymphomen. Aufgrund der CD20-Expression kann eine zusätzliche Gabe von Rituximab erwogen werden.
Therapie im Rezidiv
Die Rezidivtherapie besteht in der Regel aus einer dosisintensivierten Chemotherapie (z. B. DHAP-Protokoll) gefolgt von einer Hochdosis- Chemotherapie mit autologer Stammzelltransplantation. Damit können ca. 50% der Patienten geheilt werden, wobei jedoch Patienten mit einem Frührezidiv (< 12 Monate nach Abschluss der Primärtherapie) eine schlechte Prognose aufweisen.
ZUSAMMENFASSUNG: Die meisten Hodgkin- Lymphome sind heutzutage dank risikoadaptierter Therapiestrategien heilbar. Um die Balance zwischen Effektivität und Toxizität weiter optimieren zu können, sollten diese Patienten auch weiterhin vorzugsweise im Rahmen klinischer Studien behandelt werden.

Ursprünglich erschienen:
UIM 05|2011
UIM 05|2011
Herausgeber: Univ.-Prof. Dr. Günter J. Krejs, Österreichische Gesellschaft für Innere Medizin
Publikationsdatum: 2011-06-17
Zur Ausgabe »
Publikationsdatum: 2011-06-17
Zur Ausgabe »