


























































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
CHIP/ARCH und kardiovaskuläre Medizin
4. Mai 2020
Was ist klonale Hämatopoiese?
Das Knochenmark ist ein hochproliferatives Gewebe, das täglich 1010–1012 neue Blutzellen generiert. In der hierarchischen Ordnung stellen unreife hämatopoietische Stammzellen die zelluläre Basis dieses hochproliferativen Organs dar. Genetische Veränderungen in blutbildenden Stammzellen können bösartige Bluterkrankungen wie akute Leukämien (AML) oder myeloproliferative Neoplasien (MPN) auslösen, wobei hier in der Regel eine Sequenz an genetischen Ereignissen zur Transformation der Zellen führt. Einzelne genetische Läsionen im Stammzell-Pool sind nicht selten, was durch die Tatsache verdeutlicht wird, dass bis zu 17 Mutationen pro Stammzelle pro Jahr auftreten (Lee-Six, Nature 2018). Im Jahr 2014 wurde erstmalig die altersabhängige Frequenz einzelner somatischer Mutationen im Blut gesunder Menschen nachgewiesen. Hierbei handelt es sich um Mutationen, die bereits Jahre zuvor als rekurrente Veränderungen bei myeloischen Erkrankungen wie beispielsweise AML oder MPN beschrieben wurden. Die häufigsten Mutationen bei gesunden Menschen sind in den Genen TET2, DNMT3a gefolgt von ASXL-1, TP53, JAK2 nachweisbar (Jaiswal, NEJM 2014; Genovese, NEJM 2014; Xie, Nat Med 2014). Die Allelfrequenzen variieren und zeigen, dass teilweise trotz normalen Blutwerten ein erheblicher Anteil der Blutzellen klonalen Ursprungs sein kann, ohne dass als Folge des genetischen Fehlers bereits eine Bluterkrankung ausgelöst wurde. Der Nachweis solcher Veränderungen gelingt mittels Next Generation Sequencing (NGS), wobei hier neben der Frage des Vorhandenseins eines klonalen Markers (also Nachweis einer Mutation ja oder nein) auch die Allelfrequenz bestimmt werden kann. Je sensitiver die Nachweismethode, desto höher ist der Anteil an Patienten, die eine Clonal Hematopoiesis of Indeterminate Potential (CHIP) haben (Jaiswal, Science 2017). Ein anderer Begriff für CHIP ist Age-related Clonal Hematopoiesis (ARCH). In der klinischen Routine werden diese Untersuchungen bisher nicht durchgeführt, wobei sich dies aufgrund der in den folgenden Abschnitten beobachteten Bedeutung von CHIP/ARCH in Zukunft ändern dürfte.
Was bedeutet das hinsichtlich hämatologischer Neoplasien? Das Risiko, an einer hämatologischen Neoplasie zu erkranken, ist bei gesunden Personen mit noch normalen Blutwerten, aber CHIP/ARCH signifikant erhöht. Dies wird durch eine ca. 5-fach erhöhte kumulative Inzidenz an hämatologischen Neoplasien in der CHIP/ARCH-Population nach 120 Monaten unterstrichen, wobei die krebsbedingte Mortalität sich zwischen den Kohorten nicht unterschieden hat (Jaiswal, NEJM 2014).
Der Zusammenhang zwischen CHIP/ARCH und kardiovaskulärer Medizin
Verblüffend war 2014 jedoch die Nachricht, dass trotz der nicht gesteigerten Krebs-assoziierten Mortalität die Gesamtmortalität in der CHIP/ARCH-Kohorte deutlich höher war, was primär durch eine Zunahme an kardiovaskulär bedingten Todesfällen erklärbar ist. Nach Elimination krebsbedingter Todesfälle war die Sterblichkeit in der CHIP/ARCH-Kohorte mit einer Hazard Ratio von 12,9 erhöht (Genovese, NEJM 2014). Das KHK- oder Schlaganfallrisiko war bei CHIP/ARCH-PatientInnen 2-fach höher, wobei das in seiner Bedeutung in etwa mit anderen akzeptierten kardiovaskulären Risikofaktoren wie beispielsweise Diabetes mellitus, Rauchen oder Hyperlipidämie gleichzusetzen ist (Jaiswal, NEJM 2014). Auch nach Adjustierung für andere Risikofaktoren zeigte sich der Effekt von CHIP/ARCH hinsichtlich eines erhöhten kardiovaskulären Erkrankungsrisikos und auch eines erhöhten koronaren Kalzifikations-Scores in der Computertomographie (Jaiswal, NEJM 2017). Auch für das Überleben nach TAVI (Mas-Peiro, Eur Heart J 2020) und bei Herzinsuffizienz (Dorsheimer, JAMA Cardiol 2019) konnte ein vergleichbar prognostisch ungünstiger Effekt von CHIP/ARCH-Mutationen gezeigt werden. Kausalpathogenetische Zusammenhänge können damit natürlich nicht nachgewiesen werden, da CHIP/ARCH z. B. lediglich einen Ausdruck eines akzelerierten Alterungsprozesses darstellen könnte, der bekanntermaßen über die im Alter akkumulierenden anderen Risikofaktoren zu kardiovaskulären Ereignissen führt. Erste Daten aus Tiermodellen aber suggerieren, dass die genetisch veränderten (klonalen) myeloischen Zellen an der Progression der Atherosklerose beteiligt sein können.

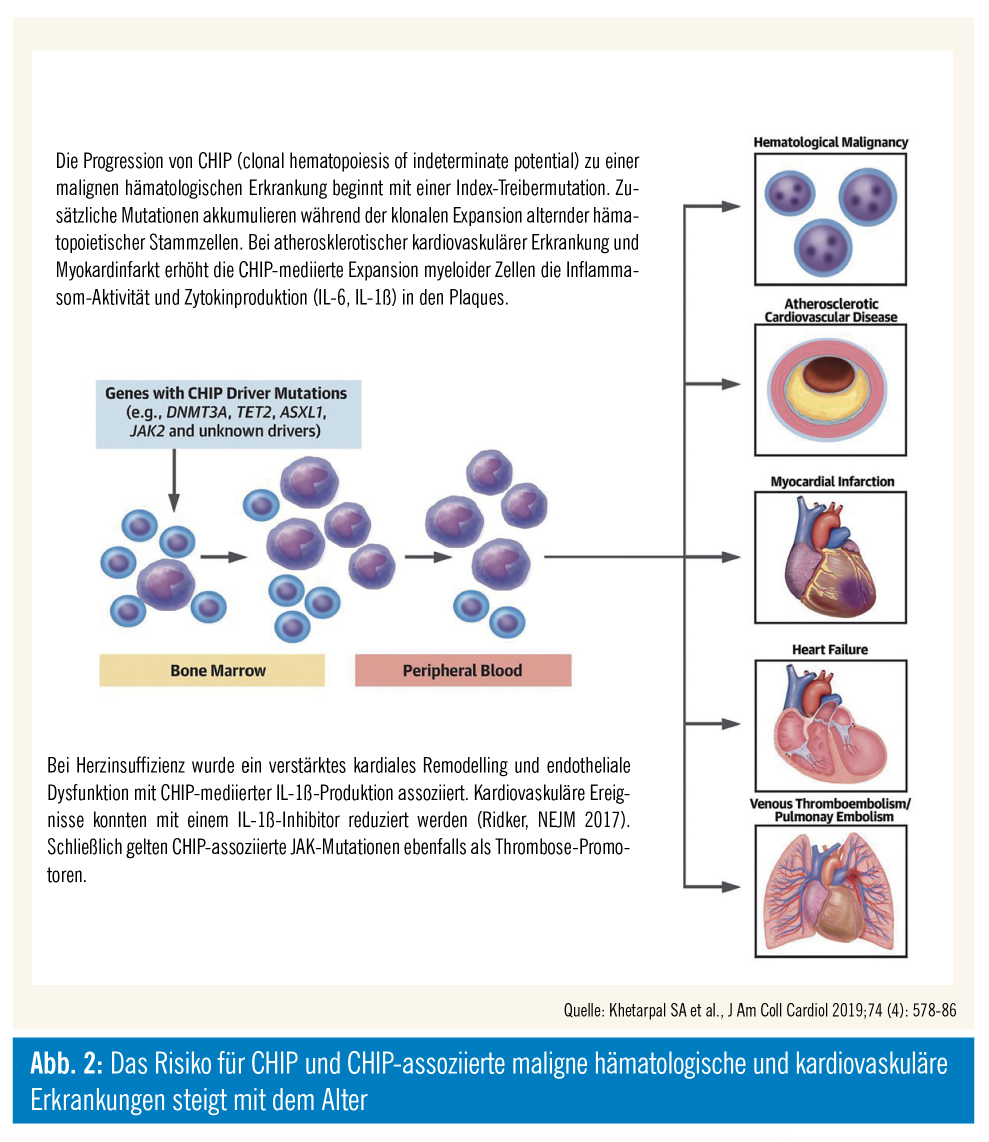
Mechanistische Modelle
Im Jahre 2012 wurde bereits in wenigen Individuen gezeigt, dass TET2-Mutationen bei älteren, sonst gesunden Individuen nachgewiesen werden können (Busqze, Nat Gen 2012). TET2 ist ein epigenetischer Regulator, der über die Metabolisierung von 5-Methylcytosin in 5-Hydroxymethylcytosin die DNA-Demethylierung und damit transkriptionelle Aktivität in Zellen fördert. In den oben bereits zitierten Arbeiten 2014 konnte dann auch verifiziert werden, dass TET2 die am häufigsten auftretende CHIP-Mutation ist. In Tiermodellen konnte nun nach kompetitiven Transplantationsmodellen durch eine 1:9-Mischung von TET-defizienter mit Wild-Typ-Blutbildung eine CHIP-artige Situation simuliert werden. Dies wurde in Mäusen getan, die aufgrund eines genetischen Defekts im LDL-Rezeptor eine Atherosklerose entwickeln. Hierbei konnte im Vergleich zur Transplantation mit ausschließlich normaler Hämatopoiese in den „CHIP-like“-Mäusen (mit kompletter oder haploinsuffizienter TET2-defizienter Hämatopoiese) eine Beschleunigung der Atherosklerose gezeigt werden (Fuster, Science 2017). Vergleichbare Daten zeigten sich in einer parallelen Arbeit, wobei hier ein weniger CHIP-relevantes Modell verwendet wurde, da die gesamte Blutbildung TET2-defizient war (Jaiswal, NEJM 2017). Interessanterweise genügte es, den genetischen Defekt in Monozyten/ Makrophagen zu induzieren, was darauf hinweist, dass diese Zellen möglicherweise atherosklerotische Plaques infiltrieren und dort eine verstärkte Entzündungsreaktion und damit entzündliche Plaqueprogression vorantreiben. Passend zu dieser Hypothese produzieren TET2-defiziente Makrophagen vermehrt Interleukin-1ß, das wiederum die Expression von P-Selektin auf Endothelzellen und damit die Rekrutierung von Entzündungszellen verstärkt. Die erhöhte IL-1ß-Produktion ist Folge einer NLRP3-Inflammasom-Aktivierung in TET-2 defizienten Tieren, was zur Cleavage von löslichem pro-IL-1ß über die Aktivierung der Caspase-1-Aktivität führt (Fuster, Science 2017). Der genaue Mechanismus der Aktivierung von NLRP3 als Folge von Mutationen im TET2-Gen ist bisher unbekannt, jedoch konnte auch in 52 epidemiologischen Studien im Rahmen des TOPMed-Programms eine Assoziation von TET2-Mutationen im Blut und erhöhten IL-1ß-Spiegeln beschrieben werden (Bick AG, Preprint 2020). Klinisch lässt sich der Zusammenhang zwischen vermehrter IL-1ß- Produktion und dem Risiko kardialer Ereignisse auch aus der CANTOS-Studie ableiten. Ein neutralisierender Antikörper gegen IL-1ß (Canakinumab) führte bei Hochrisikopatienten dosisabhängig zu einer Risikoreduktion kardialer Ereignisse (Ridker, Lancet 2017), wobei der Effekt besonders bei jenen Patienten nachweisbar war, die zeitgleich eine TET2-Mutation im Sinne einer CHIP/ARCH hatten (Svensson, Circulation 2018).
Resümee
Bei älteren Patienten gibt es einen signifikanten Anteil an kardiovaskulären Ereignissen ohne Vorliegen klassischer kardiovaskulärer Risikofaktoren (Vernon, J Am Heart Assoc 2019). Die Präexistenz von CHIP/ARCH könnte hier ein erklärender Faktor sein, der die entzündliche Pathogenese der Atherosklerose fördert. Sollte sich in prospektiven Studien verifizieren lassen, dass CHIP/ARCH-induziertes inflammatorisches Imprinting myeloischer Zellen einen validen Risikofaktor darstellt, wären in genau dieser Patientenkohorte antientzündliche Strategien evtl. von besonderes hohem Nutzen in der Primär-, aber auch der Sekundärprävention. Vorstellbar wären hier in Zukunft Medikamente, die spezifisch die NLRP3/IL-1ß-Achse hemmen, oder auch breitere antientzündliche Strategien wie beispielsweise der Einsatz von Kolchizin.
Ausgewählte Literatur
-Lee-Six H et al. Population dynamics of normal human blood inferred from somatic mutations. Nature 2018; 561: 473-478
-Jaiswal S et al. Age-Related Clonal Hematopoiesis Associated with Adverse Outcomes. N Engl J Med 2014; 371: 2488-2498
-Genovese G et al. Clonal Hematopoiesis and Blood-Cancer Risk Inferred from Blood DNA Sequence. N Engl J Med 2014; 371: 2477-2487
-Xie M et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med 2014; 20 (12): 1472-1478
-Fuster JJ et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science 2017; 355 (6327): 842-847
-Jaiswal S et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N Engl J Med 2017; 377: 111-121
-Mas-Peiro S et al. Clonal haematopoiesis in patients with degenerative aortic valve stenosis undergoing transcatheter aortic valve implantation. Eur Heart J 2020, 41 (8): 933-939
-Dorsheimer L et al. Association of Mutations Contributing to Clonal Hematopoiesis With Prognosis in Chronic Ischemic Heart Failure. JAMA Cardiol 2019; 4 (1): 25-33
-Busque L et al. Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat Genet 2012; 44 (11): 1179-1181
-Bick AG et al. Inherited Causes of Clonal Hematopoiesis of Indeterminate Potential in TOPMed Whole Genomes. https://www.biorxiv.org/content/10.1101/782748v1
-Ridker PM et al. Effect of interleukin-1 inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet 2017; 390 (10105): 1833-1842
-Svensson EC et al. TET2-Driven Clonal Hematopoiesis Predicts Enhanced Response to Canakinumab in the CANTOS Trial: An Exploratory Analysis. Circulation 2018; 138 (Suppl 1): A15111
-Vernon ST et al. ST-Segment-Elevation Myocardial Infarction (STEMI) Patients Without Standard Modifiable Cardiovascular Risk Factors – How Common Are They, and What Are Their Outcomes? J Am Heart Assoc 2019; 8 (21): e013296

AutorIn: Univ.-Prof. Dr. Dominik Wolf
Innere Medizin 5, Abteilung für Hämatologie und Onkologie, Medizinische Universität Innsbruck

AutorIn: Univ.-Prof. Dr. Axel Bauer
Innere Medizin 1, Abteilung für Kardiologie und Angiologie, Medizinische Universität Innsbruck

Ursprünglich erschienen:
SO 02|2020
SO 02|2020
Herausgeber: Univ.-Prof. Dr. Matthias Preusser, Univ.-Prof. Dr. Markus Raderer
Publikationsdatum: 2020-05-04
Zur Ausgabe »
Publikationsdatum: 2020-05-04
Zur Ausgabe »