


























































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Grundlegende genetische Mechanismen der klonalen Evolution
4. Mai 2020
Mehr als 150 Jahre nach Darwins Evolutionstheorie und infolge des bahnbrechenden technischen Fortschritts durch massiv-parallele Sequenzierung (NGS) sowie Einzelzell-Sequenzierung zeigt sich nun, dass „survival of the fittest“ nicht nur in der Evolution der Organismen, sondern auch in der Evolution der Tumorerkrankungen eine zentrale Rolle spielt.
Zu Beginn der Entwicklung eines Organismus gibt es nur eine einzige Genomausstattung. Durch unterschiedliche Regulationsmechanismen werden Gene ein- und ausgeschalten, wodurch unterschiedliche Gewebearten sowie Organfunktionen ausgebildet werden. Im Rahmen der Entwicklung finden in den Zellen unvermeidlich neu auftretende Veränderungen des Genoms (somatische Mutationen) statt. Die meisten dieser Veränderungen sind neutral, d. h., sie treten in nicht-regulatorischen oder nicht-kodierenden genomischen Abschnitten auf und haben somit keinen Einfluss auf den Zellphänotyp. Ist jedoch eine essenzielle Funktion von einer solchen Mutation betroffen, kann eine Zelle einen Selektionsvorteil oder -nachteil gegenüber den anderen Zellen des gleichen Organs erhalten. Bei Tumoren kann z. B. eine beschleunigte Proliferation zu einer klonalen Expansion führen. Dies spielt eine wesentliche Rolle z. B. bei der klonalen Hämatopoese bis hin zur Entstehung einer bösartigen Tumorerkrankung.
Klonale Evolution
Verschiedene neue massiv-parallele Sequenziermethoden erlauben mittlerweile die Detektion somatischer Veränderungen in einem kleinen Anteil von Zellen (targeted oder whole exome sequencing: Cutoff 0,01 % bzw. 0,07 % Variante Allel-Frequenz, VAF). Dadurch kann die klonale Evolution eines Tumors besser nachvollzogen und v. a. verstanden werden.2, 3 Allerdings birgt der Einsatz neuer Technologien auch neue Herausforderungen. Die Schwierigkeit liegt in der Beurteilung, ob ein technisch nachgewiesener Klon tatsächlich eine funktionelle Relevanz aufzeigt. So stellt sich z. B. beim Nachweis von klonalen hämatopoetischen Veränderungen die Frage, ob bzw. wann eine Indikation zu einer Therapie notwendig sein könnte, um die Transformation in eine hämatologische Neoplasie zu verhindern. Wo liegt der Unterschied zwischen einer klonalen Hämatopoese, die keine relevanten Konsequenzen hat, und einer klonalen Hämatopoese als Vorstufe einer malignen Erkrankung?
Formen der klonalen Evolution
Die klonale Evolution ist ein aktiver Prozess, der das Genom durch ein dynamisches Zusammenspiel neu auftretender genetischer und epigenetischer Veränderungen unter evolutionärer Selektion kontinuierlich prägt. Die Tumorheterogenität basiert auf mehrstufigen genetischen Veränderungen, die zu einem Tumoraggregat aus mehreren Zellpopulationen führen. Darüber hinaus ist die klonale Evolution maßgeblich am Krankheitsphänotyp sowie an der Progression bzw. der Regression der Tumorerkrankung beteiligt. Molekulargenetische Studien zeigten beispielsweise, dass sich eine AML im Krankheitsverlauf vom Zeitpunkt der Diagnose bis zum Rückfall in ihrer genetischen Zusammensetzung stark verändert. Unterschiedliche Klone können dabei im Krankheitsverlauf auch verschiedene klonale Evolutionen durchlaufen.4
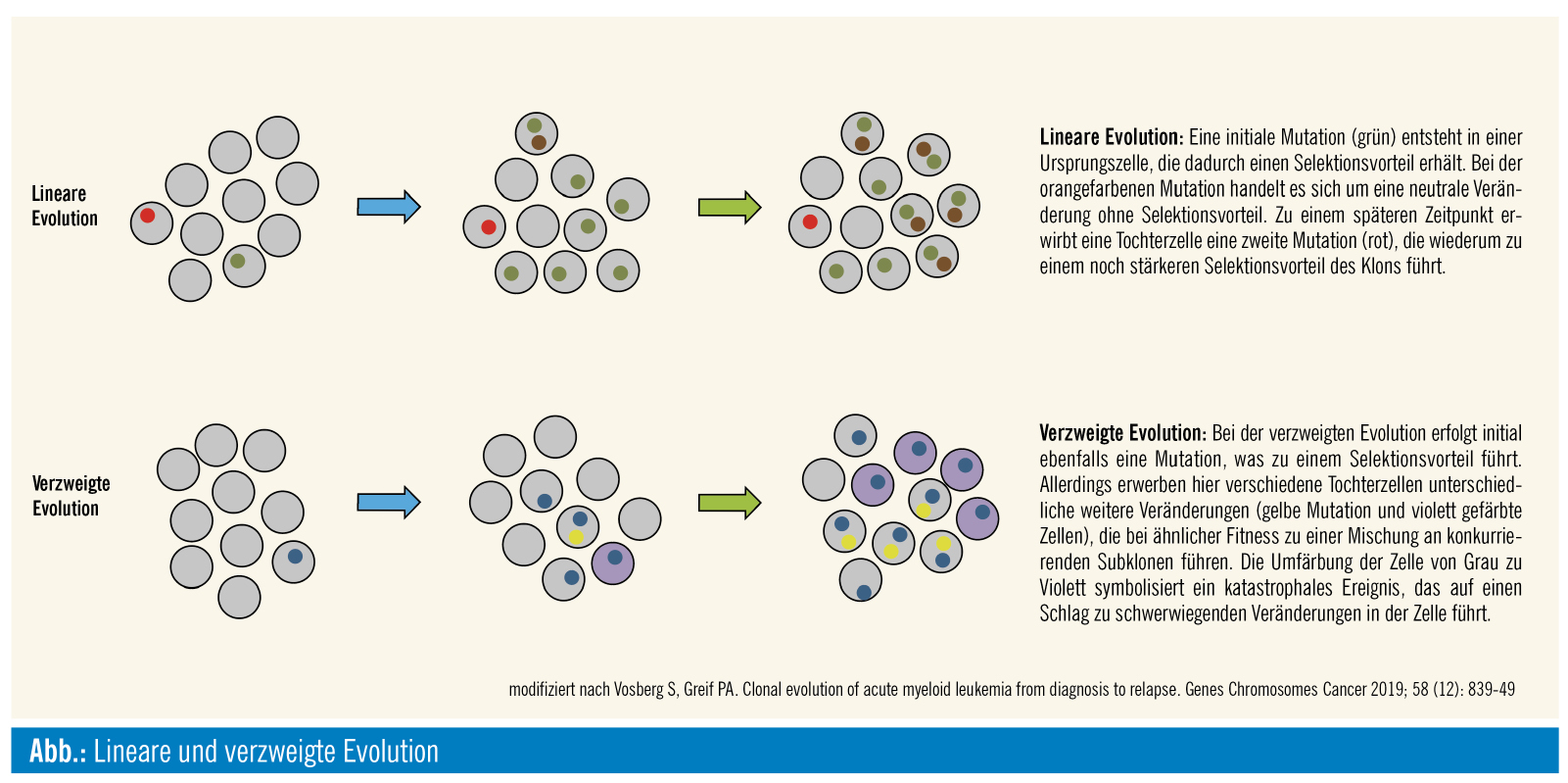
Lineare und verzweigte Evolution (Abb.): Bei einer linearen Evolution erwirbt eine Zelle eine Mutation und erhält dadurch ggf. einen Selektionsvorteil. Diese Veränderung gibt sie in der Folge an alle Tochterzellen weiter. Bei der genetischen Diagnostik dieser Erkrankung kann man sowohl bei der initialen Diagnose als auch bei einem Rückfall der Erkrankung dieselbe Mutation nachweisen. Meist treten im weiteren Verlauf in unterschiedlichen Tochterzellen unterschiedliche weitere Mutationen auf, es entstehen Subklone, die den Ausgangsklon bzw. andere Subklone verdrängen. Man spricht von einer multilinearen oder verzweigten Evolution (branched evolution). Im Modell dieser Evolutionsform können mehrere Klone koexistieren und sich abhängig u. a. von der Therapie unterschiedlich weiterentwickeln. Diese komplexe genetische Transformation führt dazu, dass sich unterschiedliche Mutationsmuster im Rahmen der initialen Diagnose und bei der rezidivierten Erkrankung nachweisen lassen.
Neutrale Evolution: Das Gegenteil einer verzweigten Evolution ist die neutrale Evolution, bei der mehrere Klone gleichzeitig existieren, wobei keiner der Klone einen Selektionsvorteil oder eine erhöhte Fitness aufweist.
Eine Evolution kann auch durch ein einmaliges katastrophales Ereignis, das zu einer erheblichen genetischen Veränderung im Tumor führt, ausgelöst werden. Meist wird so eine punktuelle Evolution von einer starken Selektion zugunsten eines auf diese Weise neu entstandenen Klons gefördert. In einem System, in dem kein evolutionärer Druck vorliegen würde, würde eine Zelle dagegen aller Wahrscheinlichkeit nach eine lineare Evolution anstreben und stetig ihre Fitness verbessern.4–6
Mutations-Reihenfolge
Die Reihenfolge des Auftretens einer Mutation in der klonalen Evolution: Studien bei myelodysplastischen Syndromen (MDS) und myeloproliferativen Neoplasien (MPN) zeigten, dass auch die Reihenfolge des Auftretens von Mutationen den Phänotyp und den Verlauf einer Erkrankung, das Therapieansprechen sowie die weitere klonale Evolution stark beeinflusst. Die JAK2V617F-Mutation findet sich bei MPN mit einer hohen Prävalenz. Allerdings zeigen 10 % aller MPN eine zweite Mutation im TET2-Gen. Hier spielt es eine wesentliche Rolle, welche der beiden Mutationen zuerst auftritt. Patienten mit JAK2V617F als Erstmutation entwickeln eher eine Polycythaemia vera als eine essenzielle Thrombozythämie und zeigen ein erhöhtes Thromboserisiko im Vergleich zu Patienten, die zuerst eine Mutation im TET2-Gen aufweisen.7
Selektionsdruck
Der Einfluss des Selektionsdrucks auf die maligne Transformation einer mutierten Zelle: Von großer Bedeutung in der klonalen Evolution sind der selektive Druck sowie ein Fitnessvorteil eines Klons gegenüber einem anderen. Ein Selektionsdruck kann durch eine altersbedingte Zunahme an somatischen Mutationen in Genen wie TET2, DNMT3A, JAK2, ASXL1, SF3B1 und TP53 verursacht werden. Die betroffenen Personen sind asymptomatisch, zeigen jedoch eine erhöhte Neigung zur Ausbildung einer hämatologischen Erkrankung.2, 3, 8, 9 Unterschiedliche Begriffe wurden gewählt, um diesen Status zu beschreiben: Age-related clonal hematopoesis (ARCH) und clonal hematopoesis of intermediate potential (CHIP) definieren sich durch den technischen Nachweis einer somatischen Variante (≥ 2 % VAF) ohne Zytopenie, während idiopathic cytopenias of undetermined significance (ICUS) eine milde Zytopenie ohne nachweisbare somatische Varianten zeigen. Die clonal cytopenias of undetermined significance (CCUS) sind durch den Nachweis einer somatischen Variante und einer Zytopenie bei normaler Knochenmarkzytologie gekennzeichnet.10 Neben dem natürlichen Druck, den die Zelle mit zunehmendem Alter erfährt, gibt es auch Selektionsvorteile aufgrund exogener Stressfaktoren wie Bestrahlung oder Chemotherapie. Dieser exogene Druck ist ein richtungsgebender Faktor für die klonale Evolution und ist der natürlichen Selektion übergeordnet. Studien mit Patienten nach einer Chemotherapie (als Teil der Behandlung einer nicht-hämatologischen Erkrankung) zeigten nicht nur die altersbedingten Veränderungen, sondern auch Mutationen in Genen wie TP53, PPM1D, ATM und CHEK2, welche eine wichtige Rolle bei der DNA-Kontrolle und DNA-Reparatur (DNA damage response) spielen.11–13
Diese Mutationen weisen auf einen exogenen Selektionsdruck hin, der zur Entwicklung von DNA-damage response resistenten Klonen führt. Allerdings geht aus diesen Studien hervor, dass Klone z. B. mit TP53-Mutation bereits in den präleukämischen Stadien zu finden waren und erst aufgrund der positiven Selektion wesentlich zur Krankheitsprogression beitragen.14
Epigenetische Faktoren
Zusammenspiel von genetischen und epigenetischen Faktoren: Epigenetische Prozesse und die dadurch verursachten Änderungen in der Chromatinorganisation spielen eine zentrale Rolle in der Genregulation und leisten einen erheblichen Beitrag zur klonalen Evolution. Bemerkenswert ist, dass Leukämien ebenso wie andere hämatologische Erkrankungen eine weitaus höhere Zahl an epigenetischen als an genetischen Veränderungen aufweisen.15–20 Methylomanalysen von unterschiedlichen Leukämien weisen darauf hin, dass die Tumorzellen bei einem Rezidiv eher hypomethyliert sind.21 Hypermethylierte akute myeloische Leukämie (AML), chronische lymphatische Leukämie (CLL) und chronische myeloische Leukämie (CML) sind mit einem weitaus aggressiveren und klinisch ungünstigeren Verlauf assoziiert.22–25 Der genaue Zusammenhang zwischen epigenetischer Regulation und spezifischen genetischen Veränderungen ist allerdings noch weitgehend unklar.
Der technologische Fortschritt erlaubt tiefe Einblicke in die Tumorheterogenität und die damit verbundene klonale Evolution. Zudem zeigt sich, dass im Rahmen einer malignen Erkrankung die genetischen Veränderungen von Beginn bis zu Progression und Rezidiv stark variieren können, wobei auch die Reihenfolge der auftretenden Mutationen einen starken Einfluss auf den Krankheitsphänotyp haben kann. Basierend auf diesem Wissen könnten zukünftig Mutationen, die im Verlauf der klonalen Evolution auftreten, als Biomarker dienen und bei Therapieentscheidungen ausschlaggebend sein.
1Darwin CR. Über die Entstehung der Arten. Übersetzung der 6. Auflage (2016). Altenmünster, Deutschland: Jazzbee Verlag
2Coombs CC et al. Therapy-Related Clonal Hematopoiesis in Patients with Non-hematologic Cancers Is Common and Associated with Adverse Clinical Outcomes. Cell Stem Cell 2017; 21 (3): 374-82.e4
3Jaiswal S et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med 2014; 371 (26): 2488-98
4Vosberg S, Greif PA. Clonal evolution of acute myeloid leukemia from diagnosis to relapse. Genes Chromosomes Cancer 2019; 58 (12): 839-49
5Davis A, Gao R, Navin N. Tumor evolution: Linear, branching, neutral or punctuated? Biochim Biophys Acta Rev Cancer 2017; 1867 (2): 151-61
6Gerlinger M et al. Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nat Genet 2014; 46 (3): 225-33
7Ortmann CA et al. Effect of mutation order on myeloproliferative neoplasms. N Engl J Med 2015; 372 (7): 601-12
8Lindsley RC et al. Prognostic Mutations in Myelodysplastic Syndrome after Stem-Cell Transplantation. N Engl J Med 2017; 376 (6): 536-47
9Genovese G et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med 2014; 371 (26): 2477-87
10Bejar R. CHIP, ICUS, CCUS and other four-letter words. Leukemia 2017; 31 (9): 1869-71
11Kastan MB, Bartek J. Cell-cycle checkpoints and cancer. Nature 2004; 432 (7015): 316-23
12Kastenhuber ER, Lowe SW. Putting p53 in Context. Cell 2017; 170 (6): 1062-78
13Kleiblova P et al. Gain-of-function mutations of PPM1D/Wip1 impair the p53-dependent G1 checkpoint. J Cell Biol 2013; 201 (4): 511-21
14Wong TN et al. Role of TP53 mutations in the origin and evolution of therapy-related acute myeloid leukaemia. Nature 2015; 518 (7540): 552-5
15Kulis M et al. Epigenomic analysis detects widespread gene-body DNA hypomethylation in chronic lymphocytic leukemia. Nat Genet 2012; 44 (11): 1236-42
16Figueroa ME et al. DNA methylation signatures identify biologically distinct subtypes in acute myeloid leukemia. Cancer Cell 2010; 17 (1): 13-27
17Milani L et al. DNA methylation for subtype classification and prediction of treatment outcome in patients with childhood acute lymphoblastic leukemia. Blood 2010; 115 (6): 1214-25
18Geng H et al. Integrative epigenomic analysis identifies biomarkers and therapeutic targets in adult B-acute lymphoblastic leukemia. Cancer Discov 2012; 2 (11): 1004-23
19Li S et al. Distinct evolution and dynamics of epigenetic and genetic heterogeneity in acute myeloid leukemia. Nat Med 2016; 22 (7): 792-9
20Pan H et al. Epigenomic evolution in diffuse large B-cell lymphomas. Nat Commun 2015; 6: 6921
21Sandoval J et al. Genome-wide DNA methylation profiling predicts relapse in childhood B-cell acute lymphoblastic leukaemia. Br J Haematol 2013; 160 (3): 406-9
22Landau DA et al. Locally disordered methylation forms the basis of intratumor methylome variation in chronic lymphocytic leukemia. Cancer Cell 2014; 26 (6): 813-25
23Oakes CC et al. DNA methylation dynamics during B cell maturation underlie a continuum of disease phenotypes in chronic lymphocytic leukemia. Nat Genet 2016; 48 (3): 253-64
24Heller G et al. Next-generation sequencing identifies major DNA methylation changes during progression of Ph+ chronic myeloid leukemia. Leukemia 2016; 30 (9): 1861-8
25Li S, Mason CE, Melnick A. Genetic and epigenetic heterogeneity in acute myeloid leukemia. Curr Opin Genet Dev 2016; 36: 100-6

AutorIn: Emina Jukic, PhD MSc
Bereichsleitung Tumorgenetik Bereichsleitung Postnatale Zytogenetik Institut für Humangenetik Zentrum Medizinische Genetik Innsbruck Medizinische Universität Innsbruck

Ursprünglich erschienen:
SO 02|2020
SO 02|2020
Herausgeber: Univ.-Prof. Dr. Matthias Preusser, Univ.-Prof. Dr. Markus Raderer
Publikationsdatum: 2020-05-04
Zur Ausgabe »
Publikationsdatum: 2020-05-04
Zur Ausgabe »