


























































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
CKD: Hot Topics zur kardiovaskulären Kalzifikation und CKD-MBD
27. März 2019
Nachlese zum ERA-EDTA 2018
Der vorliegende Artikel stellt ein Update und eine Zusammenfassung ausgewählter Neuigkeiten zur kardiovaskulären Kalzifikation bei CKD sowie der Behandlung der CKD-MBD dar, die im Rahmen der 55. Jahrestagung der ERA-EDTA vergangenen Jahres in Kopenhagen präsentiert wurden.
Pathophysiologische Schlüsselaspekte der Mediaverkalkung bei CKD
Vaskuläre Kalzifizierung, d. h. die pathologische Ablagerung von Mineralsalzen in Gefäßen, ist keine umschriebene singuläre Entität, sondern kann sich in mehreren Formen manifestieren: als Intimaverkalkung im Sinne der Verkalkung atheromatöser Plaques, als Mediaverkalkung oder auch gemischt mit beiden Formen gleichzeitig an derselben anatomischen Stelle sowie gemischt, aber abhängig von der Gefäßlokalisation variierend. Den entscheidenden Zelltyp für die Mediaverkalkung stellt die glatte Gefäßmuskelzelle (VSMC) dar. Die Mediaverkalkung selbst ist hauptsächlich ein aktiver regulierter Prozess, der aus der Imbalance zwischen prokalzifizierenden (Kalzium, Phosphat, BMP-2, alkalische Phosphatase, Osteocalcin) und antikalzifizierenden (Matrix-GLA-Protein, Fetuin-A, BMP-7, Osteoprotegerin) systemischen und lokalen Faktoren resultiert und einige Aspekte mit der regulären Knochenbildung teilt. Der Haupttreiber der Gefäßverkalkung scheint bei CKD-Patienten die Hyperphosphatämie zu sein. Phosphat gelangt über den sog. Na/Pi-Cotransporter Pit-1 in die VSMC und induziert eine Transdifferenzierung der kontraktilen Zelle zu einem sekretorischen osteoblasten-/osteozytenähnlichen Zelltyp. Damit geht auch eine Änderung der Genexpression und Proteinsynthese einher, von Muskelmarkern wie α-Aktin oder SM22α hin zu Osteoblastenmarkern wie Osteocalcin, BMP-2 oder alkalischer Phosphatase. Von diesen Zellen sezernierte Matrixvesikel und apoptotische Körperchen bilden die Kernstrukturen („Nidus“), die mineralisieren (= extrazelluläre Ablagerung von Phosphat in Form von Hydroxyapatitnanokristallen). Neben diesen aktiven Prozessen kommt es auch passiv zur Ablagerung von Kalziumphosphat in Form sog. sekundärer Calciproteinpartikel (CPP), die Fetuin-A-gebundenes Hydroxyapatit enthalten.1 Auch die mit abnehmender Nierenfunktion beobachtbare Klotho-Defizienz (über eine verminderte Pit-1-Hemmung) sowie FGF23 und PTH (über bislang noch nicht eindeutig identifizierte Mechanismen) beeinflussen die obig beschriebene VSMC-Transdifferenzierung. Die damit einhergehenden Veränderungen der extrazellulären Matrix mit Elastinreduktion und Kollagenvermehrung aggravieren die durch die Mediaverkalkung verursachte Gefäßsteifigkeit mit allen ihren klinischen Konsequenzen (erhöhter zentraler Blutdruck, Linksventrikelhypertrophie, Herzinsuffizienz).2
Vitamin K bei CKD-Patienten
Vitamin K existiert in drei Formen: Vitamin K1 (Phyllochinon), v. a. in grünem Gemüse; Vitamin K2 (Menachinon), von dem für die vaskuläre Kalzifikation insbesondere die langkettigen Menachinone MK-7, MK-8 und MK-9 von Bedeutung sind und das sich in hoher Konzentration in fermentiertem Käse und in der japan. Spezialität Natto (fermentierte Sojabohnen, die mit Abstand reichste Vitamin K2-Quelle, besonders reich an MK-7) findet; synthetisches Vitamin K3 (Menadion). Bei Nierengesunden gibt es Hinweise für eine Reduktion der Koronarverkalkung durch höhere Menachinonzufuhr. Neben den Vitamin-K-abhängigen Gerinnungsfaktoren (II, VII, IX, X) in der Leber benötigen auch die wichtigen Kalzifizierungsinhibitoren MGP (Matrix-GLA-Protein) und GAS-6 (Growth Arrest Specific Gen 6) einen Vitamin K-abhängigen γ-Carboxylierungsschritt zur enzymatischen Aktivierung. Für diese γ-Carboxylierung ist die γ-Glutamylcarboxylase verantwortlich. Bei Patienten mit fortgeschrittener CKD lässt sich eine verminderte diätetische Vitamin-K-Zufuhr sowie eine reduzierte Aktivität der γ-Glutamylcarboxylase beobachten. Zudem greifen Vitamin-K-Antagonisten hemmend in den Vitamin-K-Recyclingzyklus und auf diese Weise auch in die γ-Carboxylierung der vaskulären Kalzifizierungsinhibitoren ein. Tierexperimentell lässt sich die Aktivität der γ-Glutamylcarboxylase in der Aorta mit der Gabe von Vitamin K2 signifikant steigern und auch die Synthese von carboxyliertem, d. h. aktivem MGP erhöhen.3, 4 Eine erste Antwort auf die Frage nach dem klinischen Nutzen einer Vitamin-K-Substitution hinsichtlich der Gefäßverkalkung bei Hämodialysepatienten erwarten wir uns aus der VitaVask-Studie. In diese prospektive, randomisierte Open-Label-Parallelgruppenstudie wurden 348 Patienten mit eine Volumenscore der Koronarverkalkung > 100 eingeschlossen und über 18 Monate in der Verumgruppe mit zusätzlich 5 mg Vitamin K1 oral 3 x/Woche zur Dialyse behandelt. Der primäre Endpunkt der Studie stellt die computertomografisch gemessene Progression der Aorten- und Koronarverkalkung nach 12 bzw. 18 Monaten dar; sekundäre Endpunkte umfassen die Gesamtmortalität und kardiovaskuläre Ereignisse (MACE) nach 3 bzw. 5 Jahren.5
Neues zur Phosphatbindertherapie?
Der Einsatz von Phosphatbindern in der Therapie der CKD-MBD basiert auf zwei Überlegungen: zum einen aus pathophysiologischen Gründen, nämlich der Beeinflussung der Hyperphosphatämie als Trigger für die Entwicklung des sekundären Hyperparathyreoidismus und der vaskulären Kalzifikation; zum anderen aufgrund mehrerer Beobachtungsstudien, die einerseits in verschiedenen Dialysepopulationen immer wieder eine Assoziation zwischen der Höhe des Serumphosphatspiegels und der Gesamtmortalität sowie kardiovaskulären Mortalität zeigten6, 7, andererseits eine Mortalitätsreduktion bei Patienten unter Phosphatbindertherapie nahelegten8, 9. Dementsprechend empfehlen die 2017 aktualisierten KDIGO-CKD-MBD-Richtlinien, bei Patienten mit CKD G3a–G5D mit Hyperphosphatämie die Serumphosphatkonzentration Richtung Normalwert zu senken. Im Hinblick auf die Gefäßkalzifikation und eine erhöhte externe Kalziumzufuhr gipfelt die Phosphatbinderdiskussion in der entscheidenden hamletischen Frage, ob in der Praxis nun ein kalziumhaltiger oder kalziumfreier Phosphatbinder verwendet werden soll. Im Vergleich zum kalziumfreien Sevelamer kommt es bereits ab einer Zufuhr von 1,2 g elementaren Kalziums pro Tag über 26 bzw. 52 Wochen zu einer signifikanten Progression der koronaren und aortalen Verkalkung.10 Hinsichtlich der Gesamtmortalität und kardiovaskulären Mortalität fand sich jedoch in der größten randomisiert-kontrollierten Studie zu dieser Fragestellung mit > 2.000 Patienten kein Unterschied zwischen Sevelamer und kalziumhaltigem Phosphatbinder.11 Trotz hauptsächlich inkonklusiver und negativer randomisierter kontrollierter Einzelstudien weisen zwei Metaanalysen der letzten 5 Jahre auf eine mögliche Mortalitätsreduktion bei Verwendung kalziumfreier Phosphatbinder hin.12, 13 Dementsprechend empfehlen auch die KDIGO-Richtlinien, kalziumhaltige Phosphatbinder möglichst restriktiv einzusetzen. Als Alternativen stehen uns mehrere Möglichkeiten zur Verfügung: Sevelamercarbonat, Colestilan, Lanthancarbonat, Eisencitrat, Eisenoxyhydroxid oder Magnesiumcarbonat (kombiniert mit niedrigdosiertem Kalziumacetat). Unterschiede in der Tablettenlast und das Nebenwirkungsprofil sind bei vergleichbarer Wirksamkeit für die Wahl des jeweiligen Präparats entscheidend.
Kalzimimetika und/oder Vitamin-D-Rezeptor-Aktivatoren (VDRA)
Neben den Phosphatbindern zählen auch Kalzimimetika (Cinacalcet, Etelcalcetid) und VDRA (Calcitriol, Alfacalcidol, Paricalcitol) zum Armamentarium in der Behandlung der CKD-MBD. Beide senken zwar die PTH-Spiegel und fördern die Expression von Kalzium-Sensing-Rezeptoren in den Nebenschilddrüsenzellen, unterschieden sich jedoch grundlegend im Hinblick auf Serumkalzium-, Serumphosphat- und FGF-23-Konzentration. Während Kalzimimetika diese senken, werden sie von VDRA tendenziell erhöht. VDRA in pharmakologischen Dosen hemmen das kardiale und renale Renin-Angiotensin-Aldosteron-System, verbessern möglicherweise Endothelfunktion und Proteinurie und mindern den Knochenturnover. Beobachtungsstudien weisen auf einen Mortalitätsbenefit hin, randomisiert-kontrollierte Studien dazu fehlen. Kalzimimetika erhöhen die Erfolgsrate, die biochemischen Parameter PTH, Kalzium, Phosphat und FGF-23 in den gewünschten Zielbereich zu korrigieren, scheinen die Gefäßverkalkung abzuschwächen, reduzieren den Knochenturnover, steigern die Knochendichte und reduzieren die Größe hyperplastischer Nebenschilddrüsen. Zudem senken sie die Parathyreoidektomiehäufigkeit, die Rate an unkontrolliertem Hyperparathyreoidismus und die Frakturhäufigkeit. Was die Mortalität betrifft, fand eine 18 Studien umfassende Metaanalyse (die natürlich von EVOLVE dominiert wurde) keinen Nutzen. Das parenterale Zweitgenerationskalzimimetikum Etelcalcetid erwies sich im direkten Vergleich mit Cinacalcet hinsichtlich der PTH-Senkung als effektiver, die gastrointestinale Nebenwirkungsrate war (zumindest in der Frühphase) vergleichbar.
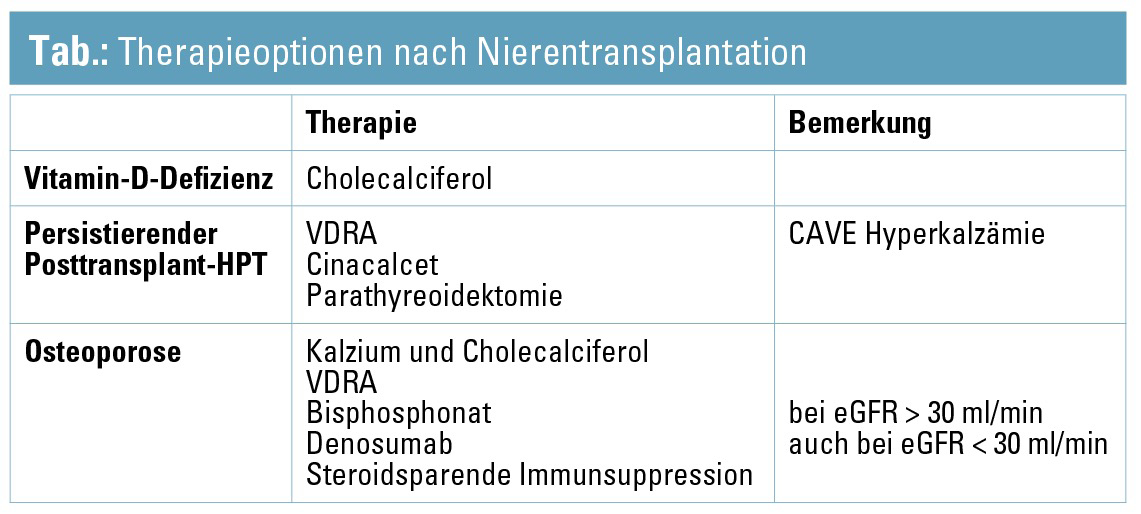
Knochenveränderungen nach Nierentransplantation
Nach Nierentransplantation können folgende histomorphometrische bzw. histologische Knochenveränderungen auftreten: steroidinduzierte Osteopenie/-porose, Osteitis fibrosa bei persistierendem Posttransplanthyperparathyreoidismus, Osteomalazie bei Vitamin-D-Defizienz, adyname Knochenerkrankung, begünstigt durch den Knochenumsatz bremsende oder hemmende Therapien (Bisphosphonate, Parathyreoidektomie, VDRA). In der frühen Posttransplantphase steigt das Frakturrisiko im Vergleich zu gelisteten Dialysepatienten um ca. 34 % an, nimmt nachfolgend dann wieder ab und sinkt nach etwa 600 Tagen nach Transplantation unter jenes der Dialysepatienten ab.14 Die möglichen Therapieoptionen sind in der Tabelle zusammengefasst. Unter Bisphosphonaten kommt es im ersten Jahr nach Transplantation zu einer leichten Zunahme der Knochendichte sowohl am Schenkelhals (+ 6 %) als auch in der Lendenwirbelsäule (+ 7,4 %).15 Auch für Denosumab konnte im ersten Posttransplantjahr eine signifikante Zunahme der Knochendichte an Schenkelhals, Hüfte und Lendenwirbelsäule nachgewiesen werden.16
RESÜMEE: Während in der Pathophysiologie der vaskulären Kalzifikation bei Patienten mit CKD einige neue Erkenntnisse gewonnen wurden, ist die optimale Präventions- und Therapiestrategie für selbige noch nicht gefunden.
1 Vervloet M., Kidney Int 2017
2 Briet M., Cli Sci 2012
3 Kaesler N., Kidney Int 2016
4 Scheiber D., Nutrients 2015
5 Krueger T., NDT 2014
6 Kalantar-Zadeh K., Kidney Int 2006
7 Tentori F., AJKD 2008
8 Isakova T., JASN 2009
9 Cannata-Andia J.B., Kidney Int 2013
10 Chertow G., Kidney Int 2002
11 Suki W.N., Kidney Int 2007
12 Jamal S.A., Lancet 2013
13 Patel L., CJASN 2016
14 Vautour L.M., Osteoporos Int 2004
15 Toth-Manikowski S.M., Clin Transplant 2016
16 Bonami M., AJT 2016

OA Priv.-Doz. Dr. Emanuel Zitt
Innere Medizin III (Nephrologie und Dialyse), Akademisches LehrkrankenhausLKH Feldkirch

Ursprünglich erschienen:
Neph 01|2019
Neph 01|2019
Herausgeber: Österreichische Gesellschaft für Nephrologie, Univ.-Prof. Dr. Rainer Oberbauer, Klinische Abteilung für Nephrologie und Dialyse, Universitätsklinik für Innere Medizin III, Medizinische Universität Wien
Publikationsdatum: 2019-03-26
Zur Ausgabe »
Publikationsdatum: 2019-03-26
Zur Ausgabe »