
























































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
ZNS-Tumoren bei Kindern und Jugendlichen
29. Juni 2012

Epidemiologie
Primäre ZNS-Tumoren stellen im Kindesalter mit 25 % aller Neoplasien nach Leukämien die zweitgrößte Gruppe maligner Erkrankungen und die häufigsten soliden Tumoren dar1. Bei ZNS-Tumoren im Kindesalter werden auch histologisch gutartige Tumoren zu den malignen Neubildungen gezählt, da die „Malignität“ des Tumors sich in einer Region wie dem ZNS nicht nur an histologischen Kriterien, sondern auch an der ungünstigen Lokalisation zeigen kann.
ZNS-Tumoren bei Kindern sind sehr heterogen in Hinblick auf Histologie, Molekularbiologie, Lokalisation und Wachstumsform. Altersgipfel bestehen zwar für bestimmte Entitäten, jedoch nicht für die Erkrankungshäufigkeit insgesamt. Hirntumoren können sogar bereits intrauterin auftreten. Der Anteil der Geschlechter ist je nach Tumorart unterschiedlich, Knaben sind insgesamt aber häufiger betroffen (1,3 : 1).
Im Österreichischen Hirntumorregister (Austrian Brain Tumor Registry, Wöhrer et al., unpublished data) werden jährlich ca. 90 Kinder- und Jugendliche mit histologisch gesicherten ZNS-Tumoren neu erfasst.
Mehrere genetische Erkrankungen können zur Entstehung von ZNS-Tumoren prädisponieren, zahlenmäßig besonders wichtig sind das erhöhte Risiko für ZNS-Tumoren bei Kindern mit Neurofibromatose Typ 1 und tuberöser Hirnsklerose.
Die relative Häufigkeit einzelner ZNS-Tumorentitäten unterscheidet sich erheblich von jenem erwachsener PatientInnen. Bei Kindern sind die niedriggradigen Gliome, vor allem pilozytische Astrozytome, mit ca. 40 % aller ZNS-Tumoren die häufigste Entität, gefolgt von der Gruppe der so genannten embryonalen Tumoren (Medulloblastom, ZNS-primitiver neuroektodermaler Tumor und atypischer teratoid rhabdoider Tumor) sowie Ependymomen und Kraniopharyngeomen.
Bei Erwachsenen machen Meningeome ein Drittel aller Hirntumoren aus, gefolgt von Glioblastomen als nächsthäufigster Entität. Bei Kindern hingegen werden Meningeome nur vereinzelt diagnostiziert. Auch die bei Erwachsenen häufigen Metastasen von Tu-moren außerhalb des ZNS kommen bei Kindern nur sehr selten vor.
Bei Kindern sind außerdem im Gegensatz zu Erwachsenen fast die Hälfte der Tumoren infratentoriell (Kleinhirn, IV. Ventrikel, Hirnstamm) lokalisiert.
Von den supratentoriellen Tumoren entstehen wiederum mehr als die Hälfte in der Mittellinie und nur ein kleinerer Teil in den Großhirnhemisphären. Nur in den ersten 2 bis 3 Lebensjahren finden sich gehäuft supratentorielle Tumoren.
Klassifikation nach Lokalisation
Histologisch gleichartige Tumoren haben bei Kindern in Abhängigkeit von der Lokalisation oft eine unterschiedliche Prognose. So kann ein pilozytisches Astrozytom, der über alle Altersgruppen häufigste ZNS-Tumor bei Kindern, operativ gut zugänglich im Kleinhirn lokalisiert sein oder infiltrativ im Opticus-Chiasma-Hypothalamus-Bereich wachsen (Tab.).
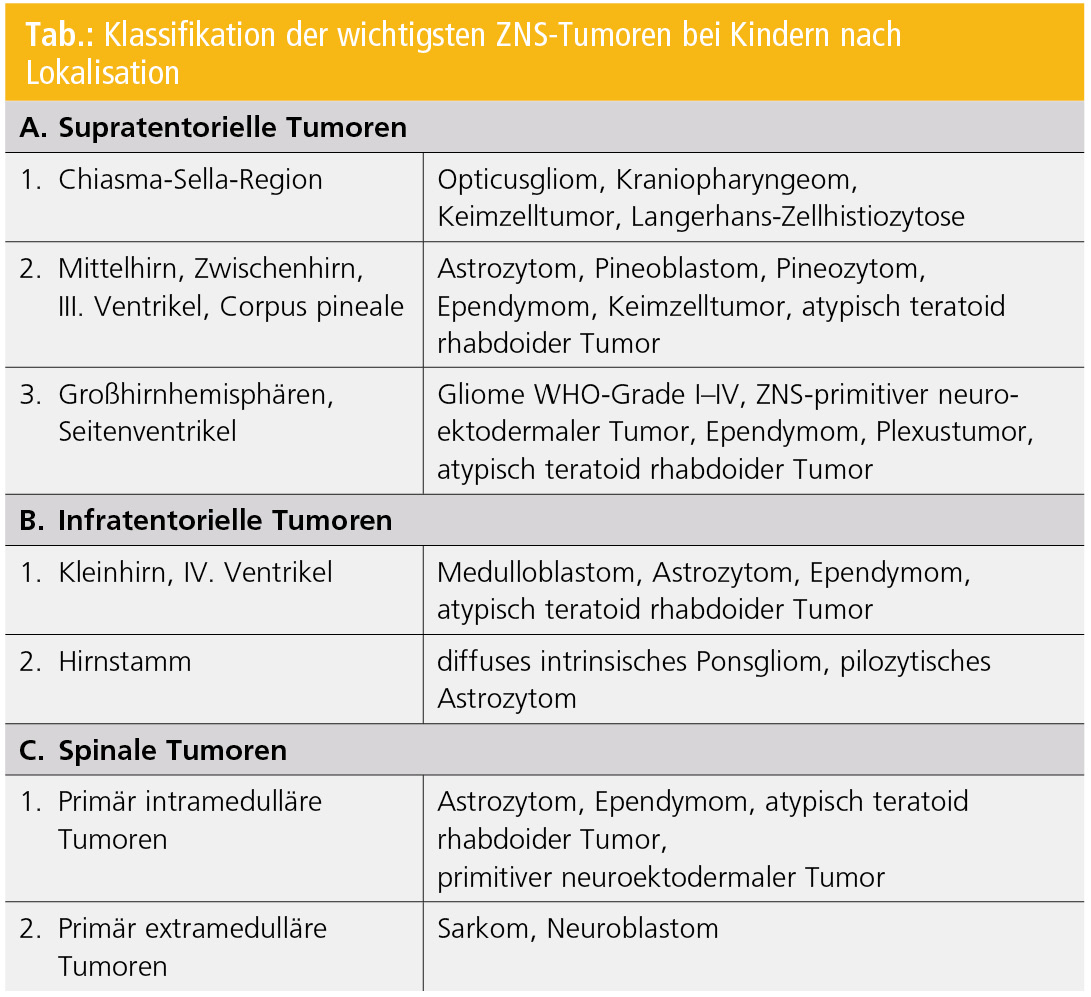
Klassifikation nach Histologie
Die Vielzahl von ZNS-Tumoren wurde ursprünglich nach histogenetischen Gesichts-punkten (putative Ursprungszellen unterschiedlichen Differenzierungsgrades) unterteilt. Diese auch prognostische Bedeutung tragende Klassifikation wird in Abständen von der WHO neuen Erkenntnissen (aus der Histomorphologie, Immunzytochemie und zunehmend Molekulargenetik) angepasst2.
Neben Unterschieden im histologischen Spektrum haben Forschungsergebnisse der letzten Jahre gezeigt, dass selbst histologisch gleichartige Tumoren, z. B. Glioblastome oder Medulloblastome, wenn sie bei Erwachsenen diagnostiziert werden, sich in ihrem genetisches Profil bzw. in der Subgruppenverteilung von Tumoren bei Kindern unterscheiden3.
Stadieneinteilung
Vor allem (aber nicht nur) embryonale ZNS-Tumoren haben eine hohe Tendenz zur leptomeningealen Aussaat. So sind z. B. Medulloblastome bei Diagnosestellung bereits in 20–40 % der Fälle metastasiert (abhängig vom Alter des/der Patienten/-in bei Diagnose). Fernmetastasen außerhalb des ZNS sind möglich, aber selten. Die Stadieneinteilung erfolgt nach der Metastasierung von M0 bis M4 (M0: keine Metastasen, M1: Tumorzellnachweis im Liquor, M2: intrakranielle Metastasen im MRT, M3: intraspinale Metastasen im MRT, M4: Metastasen außerhalb des ZNS)
Multidisziplinäres Management von ZNS-Tumoren
Seit den 1970er-Jahren haben sich die Überlebensraten von Kindern mit Hirntumoren nicht zuletzt durch Fortschritte in der Neuroradiologie, Neurochirurgie, Neuroanästhe-siologie, Strahlentherapie und Chemotherapie deutlich verbessert. Trotzdem sind ZNS-Tumoren mit ca. 32 % aller Krebstodesfälle immer noch die häufigste Krebstodesursache bei Kindern und die häufigste nicht unfallbedingte Todesursache im Kindesalter überhaupt.
Auf Grund der Heterogenität der Hirntumoren und der damit verbundenen geringen Fallzahl einer einzelnen Entität in einem Land besteht schon seit mehreren Jahrzehnten eine enge internationale Zusammenarbeit. In den letzten 30 Jahren wurden im Rahmen der „International Society of Paediatric Oncology (SIOP)“ und der „Gesellschaft für Pädiatrische Onkologie und Hämatologie (GPOH)“ multimodale Behandlungskonzepte in Therapie(optimierungs)studien systematisch weiterentwickelt. Je nach Histologie, Metastasierungsgrad und Alter des/der Patienten/-in kommen dabei Operation, Che-motherapie und Bestrahlung in zunehmend differenzierter Form zum Einsatz. Derzeit werden ca. 90 % der in Österreich an Hirntumoren erkrankten Kinder im Rahmen dieser oft komplexen internationalen Therapieprotokolle behandelt, um ein bestmögliches Überleben zu sichern.
Neben der Verbesserung der Überlebensraten ist ein weiteres Hauptziel dieser Therapiestudien die Verbesserung der Lebensqualität durch Reduktion der operations-, chemo- und strahlentherapieassoziierten Morbidität, Toxizität und Spätfolgen. Die aktuellen Hirntumorstudien der SIOP bzw. GPOH decken nahezu das komplette Spektrum der im Kindes- und Jugendalter vorkommenden Hirntumorerkrankungen ab.
Für PatientInnen bietet der Einschluss in eine Therapie(optimierungs)studie den Vorteil der Qualitätssicherung in Diagnostik (Neuropathologie, Neuroradiologie, Liquordiagnostik) und Therapie (Operation, Bestrahlung, Chemotherapie) durch die Studienzentralen mit den studienübergreifenden Referenzzentren. So können StudienpatientInnen bereits kurz nach der Diagnosestellung dem für sie nach neuestem wissenschaftlichem Stand zutreffenden Behandlungszweig der entsprechenden Therapiestudie zugeordnet werden. Ziel der risikoadaptierten Therapie, die so schonend wie möglich und nur so intensiv wie nötig durchgeführt wird, ist die Steigerung der oft beeinträchtigten Lebensqualität von Hirntumor-Überlebenden. Weitere Vorteile der Therapiestudien sind die Strukturierung von Nachsorge und Rehabilitation. Zusätzliche Ziele sind die vollständige Erfassung aller Kinder mit Hirntumoren in Österreich sowie die Bereitstellung von Tumormaterial für experimentelle Therapiestudien zur Prüfung neuer Therapieansätze und für die Grundlagenforschung4.
Um ein optimales Management zu gewährleisten, ist eine enge interdisziplinäre Zu-sammenarbeit zwischen allen medizinischen Fachbereichen, die an der Betreuung von Kindern mit Hirntumoren beteiligt sind und eine besondere Erfahrung aufweisen (NeurochirurgInnen, NeuroradiologInnen, NeuropathologInnen, KinderonkologInnen, StrahlentherapeutInnen, (Neuro-)PsychologInnen, PhysiotherapeutInnen, ErgotherapeutInnen, LogopädInnen, EndokrinologInnen, EpileptologInnen/NeuropädiaterInnen, OphthalmologInnen, OtolaryngologInnen etc.) erforderlich.
Da es sich hierbei um sehr unterschiedliche für Kinder typische Tumoren handelt, ist auf Grund der geringen Fallzahlen eine ausreichende Erfahrung naturgemäß auf wenige Zentren beschränkt. International gibt es daher einen Trend, die Behandlung dieser PatientInnen auf wenige, hochspezialisierte Zentren zu konzentrieren, die von der Operationserfahrung, intraoperativen Neuropathologie, Neuroradiologie, Strahlen- und Chemotherapie sowie neuropsychologischen Betreuung her ein bestmögliches Ergebnis gewährleisten können. Daher sollten Kinder nach Diagnose eines ZNS-Tumors so rasch wie möglich an ein kinderonkologisches Zentrum mit angegliederter, in der Behandlung von Kindern mit Hirntumoren erfahrener Neurochirurgie transferiert werden.
Therapiekonzepte der häufigsten Hirntumorentitäten
Niedriggradige Gliome: 40 % aller Hirntumoren bei Kindern- und Jugendlichen sind niedriggradige Gliome. Niedriggradige Gliome gliedern sich in WHO-Grad-I- und -II-Tumoren und umfassen mehrere Subgruppen. Die häufigste Subgruppe bei Kindern aller Altersklassen ist das pilozytische Astrozytom WHO-Grad I. In den 1970er- und 1980er-Jahren bestand die Behandlung dieser Tumoren ausschließlich in der Operation, gefolgt von einer Reoperation und/oder Strahlentherapie im Falle eines Rezidivs/Progression oder Resttumors.
Während die Resektion nach wie vor Therapie der Wahl für gut zugängliche Tumoren wie z. B. Kleinhirnastrozytome ist, hatte sich gezeigt, dass infiltrativ wachsende pilozytische Astrozytome im Opticus-Chiasma-Hypothalamus-Bereich meist nicht resektabel waren und eine schlechte Prognose hatten. Da es sich bei diesen Tumoren meist auch um sehr junge Kinder handelte, hatte die oft großflächige Strahlentherapie besonders deletäre Folgen für die kognitive Entwicklung und bedingte auch endokrinologische und vaskuläre Spätfolgen.
Seit den 1990er-Jahren werden alle Kinder mit niedriggradigen Gliomen in Österreich in internationalen Therapiestudien erfasst, zuletzt SIOP-LGG 2004. In dieser Studie wird ein genauer Therapiealgorithmus in Abhängigkeit von Resektabilität des Tumors, Lebensalter des Kindes und Vorliegen oder Fehlen einer Neurofibromatose Typ 1 als Entscheidungshilfe vorgegeben. Tumoren, die nicht resektabel sind und klinisch (fortschreitende Visusminderung, neurologische Ausfälle etc.) oder radiologisch (> 25 % Volumenzunahme) eindeutig progredient sind, werden einer auf niedriggradige Gliome abgestimmten Chemotherapie zugeführt. Mit diesem Therapieansatz ist es gelungen, die Strahlentherapie bei jungen Kindern mit großen progredienten Tumoren hinauszuzögern bzw. ganz zu vermeiden. PatientInnen ohne Neurofibromatose, die älter als 8 Jahre sind und einen relativ kleinen progredienten, nicht resektablen Tumor haben, werden lokal bestrahlt.
Neben der Rezidiv-/Progressionshäufigkeit werden auch Spätfolgen aller Art und die Lebensqualität erfasst. Neuere Untersuchungen haben gezeigt, dass ein Großteil der pilozytischen Astrozytome Veränderungen im BRAF-Gen aufweisen. Basierend auf dieser Beobachtung und präklinischen Daten, die eine Aktivität von BRAF-Inhibitoren in Xenograft-Modellen zeigten, werden derzeit international in verschiedenen Phase-I- und Phase-II-Studien BRAF-Inhibitoren und andere Signaltransduktionshemmer getestet. Da pilozytische Astrozytome histologisch eine starke vaskuläre Proliferation zeigen, werden auch verschiedene antiangiogene Medikamente in Studien erprobt.
Embryonale Tumoren – Medulloblastom, ZNS-primitiv neuroektodermaler Tumor (PNET), atypisch teratoid/rhabdoider Tumor (ATRT) – stellen die häufigsten bösartigen Tumoren bei Kindern dar. In den 1980er-Jahren gab es eine Kontroverse, ob Medulloblastome im Kleinhirn und ZNS-PNET im Großhirn unterschiedliche, lokalisationsspezifische Entitäten sind, oder ob sie unterschiedliche Manifestationen einer auf molekularer Ebene gleichartigen Tumorentwicklung sind.
Inzwischen haben Mutationsanalysen und Genexpressionsprofile gezeigt, dass nicht nur ZNS-PNET sich von Medulloblastomen unterscheiden, sondern dass es auch innerhalb der Medulloblastome mindestens 4 distinkte molekulare Untergruppen gibt, die ein unterschiedliches biologisches Verhalten und ein unterschiedliches Ansprechen auf die Behandlung zeigen. Diese neuen biologischen Erkenntnisse werden in die Risikostratifizierung der neuen Studie einfließen und die bisher verwendete klinische Risikostratifizierung (Alter 1,5 cm, primäre Metastasierung als negative Prognosefaktoren) ergänzen.
Daher gewinnen molekularbiologische Untersuchungen, für die in der Regel schockgefrorenes Frischmaterial des entnommenen Tumors erforderlich ist, zunehmend an Bedeutung und sind für die Teilnahme der PatientInnen an der neuen Studie, die 2012 starten soll, unerlässlich. In Österreich werden schon seit den späten 1980er-Jahren Kinder mit Medulloblastomen und ZNS-PNET nach den konsekutiven multimodalen HIT-Therapieoptimierungsstudien der GPOH mit zeitgemäßen radio- und chemotherapeutischen Konzepten behandelt. In der Vorläuferstudie HIT 91 lag das 10-Jahres-Überleben von Kindern über 3 Jahren mit Medulloblastomen ohne primäre Metastasierung bei 73 % und mit Metastasen bei 43 %. ZNS-PNET zeigten trotz einer geringeren initialen Metastasierung ein schlechteres Überleben.
ATRT sind hochaggressive embryonale Tumoren, die erst 1996 als eigene Entität beschrieben und 2000 in die WHO-Klassifikation aufgenommen wurden. Die Bezeichnung ATRT reflektiert das komplexe histologische Bild dieser Tumoren, die sich aus rhabdoiden Zellen mit variablen Anteilen von primitiv neuroektodermalen und mesenchymal und/oder epithelial differenzierten Zellen zusammensetzen. Die charakteristische genomische Veränderung der ATRT ist die Inaktivierung des SMARCB1-Gens (hSNF5/INI1) auf Chromosom 22q11.2, die immunhistochemisch detektiert werden kann. ATRT kommen vor allem bei Kindern vor, mit einem Altersgipfel unter 3 Jahren. In dieser Altersgruppe stellen ATRT den häufigsten malignen Hirntumor dar. ATRT treten sporadisch auf, können aber auch mit einem rhabdoiden Prädispositionssyndrom assoziiert sein. Die Prognose von Kindern mit ATRT ist ungünstig, hat sich jedoch in den letzten Jahren mit dem Einsatz intensiver Therapien einschließlich Hochdosistherapie mit autologer Knochenmarktransplantation und intrathekaler Therapie verbessert5, 6.
Hochmaligne Gliome umfassen anaplastische Astrozytome, Oligodendrogliome und Oligoastrozytome vom WHO-Grad III sowie Glioblastome und die diffusen intrinsischen Ponsgliome (WHO-Grad IV). Mit Ausnahme von Kindern unter 3 Jahren mit hochmalignen Gliomen, die eine bessere Prognose haben, sind hochmaligne Gliome schwierig zu behandeln und haben eine ungünstige Prognose. Zur Zeit besteht die Behandlung in einer multimodalen Therapie aus Operation, Chemo- und Strahlentherapie. Neuere Therapieansätze wie die Kombination der Standardtherapie mit einer auf dendritischen Zellen basierenden Impfung für Glioblastome bzw. eine Kombination mit Angiogenesehemmern werden zurzeit in mehreren Studien international und in Österreich untersucht. Aufgrund entscheidender molekularer Unterschiede von hochmaligen Gliomen bei Erwachsenen und Kindern können Studienergebnisse bei Erwachsenen, bei denen diese Tumoren häufiger vorkommen, nicht ohne weiteres auf Kinder umgelegt werden7.
Ependymome: Intrakranielle Ependymome der Grade II und III machen knapp 10 % der Hirntumoren des Kindes- und Jugendalters aus. Sie entstehen im Bereich der ependymalen Auskleidung der Ventrikel und sind im Kleinkindesalter besonders häufig. Mehr als zwei Drittel der Ependymome wachsen infratentoriell im IV. Ventrikel und zeigen hier ein biologisch ungünstiges Verhalten. Auch Ependymome verschiedener Lokalisation weisen molekulargenetisch distinkte Signaturen auf. Bei Ependymomen hat sich in mehreren Studien der Resektionsgrad als stärkster prognostischer Faktor erwiesen. Während Kinder mit einer vollständigen Resektion gefolgt von einer postoperativen Strahlentherapie eine Chance von 50–75 % aufweisen, Langzeitüberlebende zu werden, trifft das auf PatientInnen mit subtotaler Resektion nur bei 30 % zu. Der Stellenwert der Chemotherapie bei Ependymomen wird kontroversiell diskutiert, obwohl die Wirksamkeit platinbasierter Chemotherapie verschiedentlich gezeigt werden konnte. Dieser Frage wird zurzeit in einer randomisierten Phase-III-Studie (ACNS0831) in den USA nachgegangen5, 6.
Spätfolgen einer Hirntumorbehandlung
Nachdem sich die Prognose von ZNS-Tumoren bei Kindern in den letzten Jahrzehnten stetig verbessert hat und ca. zwei Drittel aller Kinder und Jugendlichen mit Hirntumoren Langzeitüberlebende werden, haben auch die Spätfolgen zunehmend an Bedeutung gewonnen.
Im Vergleich zu Tumoren außerhalb des ZNS sind die Spätfolgen für Kinder mit Hirntumoren durch den Tumor selbst, den oft über längere Zeit vor Diagnose bestehenden Hydrozephalus, operative und perioperative Komplikationen sowie die Strahlen- und Chemotherapie meist schwerwiegender. Spätfolgen nach der Bestrahlung sind vaskuläre Läsionen und Störungen der Myelinisierung. Chemotherapie mit Methotrexat kann ebenfalls die weiße Substanz der Marklager schädigen. Bestrahlung und Platinpräparate können Hördefizite verursachen. Häufig sind Beeinträchtigungen des Konzentrationsvermögens, des Kurzzeitgedächtnisses, der visuellen Wahrnehmung und des Lernvermögens. Neurokognitive Beeinträchtigungen sind bei jungen Kindern in der Regel ausgeprägter8.
Neben neurologischen Defiziten können neuroendokrinologische Funktionsstörungen durch Tumoren im Bereich der Hypophysen-Hypothalamus-Achse und deren Operation bzw. durch eine Bestrahlung dieses Bereiches ausgelöst werden. Die häufigsten strahlentherapiebedingten Endokrinopathien sind der durch Wachstumshormonmangel ausgelöste Minderwuchs, der durch das verminderte Wirbelsäulenwachstum nach kraniospinaler Bestrahlung aggraviert wird, eine primäre oder sekundäre Hypothyreose und Störungen der Pubertätsentwicklung. Eine Einschränkung der Fertilität ist nach Therapie mit Alkylantien und kraniospinaler Bestrahlung zu erwarten.
Sekundärmalignome treten bei 3–5 % der Fälle innerhalb von 5–10 Jahren nach der Behandlung auf. Zweittumoren wie Meningeome, Glioblastome und Schilddrüsenkar-zinome entstehen meist im Bereich des Strahlenfeldes mit einer Latenz von 5–40 Jahren4. Diese Spätfolgen, die die Lebensqualität der geheilten Kinder beeinträchtigen, bedürfen einer spezialisierten Langzeitnachbetreuung und stellen zunehmend auch das Forschungsfeld internationaler Studienkooperationen dar (z. B. PanCareSurfUp [PanCare Childhood and Adolescent Cancer Survivor Care and Follow-Up Studies]).
Resümee
Zusammenfassend haben Fortschritte in den bildgebenden Verfahren, den neurochirurgischen und strahlentherapeutischen Techniken und den multinationalen Chemotherapiestudien zu einer Verbesserung des Überlebens bei verschiedenen Entitäten von Hirntumoren geführt, wie z. B. dem Medulloblastom. Trotz dieser Fortschritte für einige Entitäten ist die Prognose von Kindern mit diffusen intrinsischen Ponsgliomen und hochmalignen Gliomen nach wie vor enttäuschend. Für diese, aber auch für Tumoren mit einer günstigeren Prognose liegt deshalb die Hoffnung in einer zunehmend besseren molekularen Charakterisierung und der Entwicklung zielgerichteter Therapien, die individuell und maßgeschneidert gegen spezifische Merkmale eines Tumors gerichtet sind. Neben der Verbesserung des Überlebens rückt heutzutage auch die Vermeidung von und der Umgang mit Spätfolgen der Tumoren und deren Therapie in den Vordergrund.
1 Cancer Stats Childhood Cancer – Great Britain & UK© Cancer Research UK 2010; Registered charity in England and Wales (1089464) and Scotland (SC041666); in-fo.cancerresearchuk.org/cancerstats
2 Slavc I, Tumore des Zentralnervensystems (ZNS) im Kindes- und Jugendalter. In Salzer-Muhar U et al. (Hg), Säugling, Kindheit und Jugend: MCW – Block 16, 6. Aufl. Wien 2010; Facultas. ISBN 978-3-7089-0614-0
3 Korshunov A, Remke M, Werf WT et al., Adult and pediatric medulloblastomas are genetically distinct and require different algorithms for molecular risk stratification. J Clin Oncol 2010; 28:3054–60
4 Rutkowski S et al., Hirntumoren bei Kindern und Jugendlichen. Monatsschrift Kinderheilkd 2008; 156:1165–72
5 Rutkowski S, Fleischhack G, Medulloblastome, primitive neuroektodermale Tumoren und Ependymome. Monatsschr Kinderheilkd 2008; 156:1187–93
6 Pollack I, Multidisciplinary management of childhood brain tumors: a review of outcomes, recent advances, and challenges. J Neurosurg Pediatrics 2011; 8:135–148
7 MacDonald TJ, Aguilera D, Kramm CM, Treatment of high-grade glioma in children and adolescents. Neurooncology 2011; 13(10):1049–58
8 Ellenberg L, Liu Q et al., Neurocognitive status in long-term survivors of childhood CNS malignancies: a report from the Childhood Cancer Survivor Study. Neuropsychology 2009; 23:705–17

Ursprünglich erschienen:
neuro 02|2012
neuro 02|2012

