


























































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Molekularpathologie mit Bezug auf therapierelevante Untersuchungen bei urologischen Tumoren
17. Dezember 2018
Die Mutationsspektren, epigenetischen Veränderungen und Genexpressionsprofile der meisten Tumorentitäten sind einschließlich der urologi-schen Tumoren, beschleunigt durch die Technik des „next-generation sequencing“ (NGS), bekannt.1 Das Wissen über den genetischen Bauplan von Tumoren ermöglicht die Entwicklung von zielgerichteten Therapien und fließt zunehmend in die Diagnostik ein. Eine molekularpathologische Analyse von Tumoren kann die histopathologische Diagnose unterstützen, bestimmt Prognosefaktoren, identifiziert therapeutische Zielmoleküle und Resistenzmechanismen und ermöglicht eine Stratifizierung von PatientInnen für die Wahl der optimalen Therapie.2 Bei urologischen Tumoren sind molekularpathologische Untersuchungen derzeit allerdings vorwiegend nur als Unterstützung für eine verbesserte histopathologische und zytologische Diagnose in Verwendung. Molekulare Marker für die Stratifizierung von Patienten für zielgerichtete Therapien haben mit Ausnahme der PD-L1-Expression für die Immun-Checkpoint-Inhibitor-Therapie beim Blasenkarzinom noch nicht Einzug in den klinischen Alltag gehalten.3 Im Folgenden wird eine Übersicht von derzeit bei urologischen Tumoren in der Routinediagnostik verwendeten molekularpathologischen Analysen gegeben.
Nierenzellkarzinom
Die histopathologische Klassifikation von Nierenzellkarzinomen, welche Subtypen wie das klarzellige (80 %), papilläre (10 %) und chromophobe (5 %) Karzinom und andere seltenere Histologien umfasst, gibt prognostische Informationen. Molekulapathologische Untersuchungen können in schwierigen Fällen die histologische Beurteilung bei der Tumorklassifikation unterstützen (Tab.). Zum Beispiel weist das klarzellige Nierenzellkarzinom eine Deletion im kurzen Arm von Chromosom 3 auf. Diese Deletion kann mittels Fluoreszenz-in-situ-Hybridisierung (FISH) am histologischen Schnitt nachgewiesen werden. Der kurze Arm von Chromosom 3 enthält das Tumorsuppressorgen Von Hippel Lindau (VHL). Es kodiert für eine E3-Ubiquitin-Ligase, welche den Transkriptionsfaktor HIF1-α (Hypoxia-inducible Factor 1-alpha) für den Abbau im Proteasom markiert. Bei einem Ausfall von VHL ist die HIF1-α-Aktivität erhöht, und die Expression von proangiogenetischen Proteinen wird stimuliert.4 Dieser Mechanismus ist die Grundlage für die Wirksamkeit von VEGFR-(Vascular endothelial growth factor receptor-)Inhibitoren, wie z. B. Sorafenib oder Sunitinib, in der Therapie des Nierenzellkarzinoms.
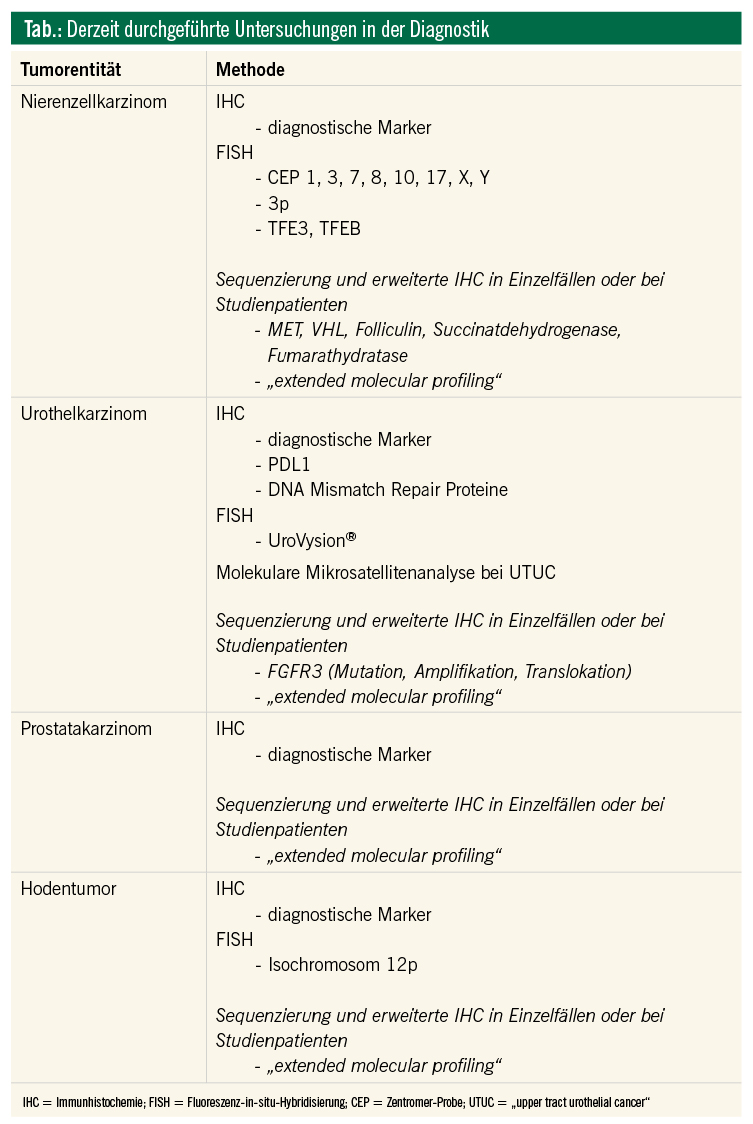
Ähnlich dem klarzelligen Karzinom weisen andere Subtypen des Nierenzellkarzinoms Chromosomenaberrationen auf, welche für die Tumorklassifikation mittels FISH nachgewiesen werden können. Papilläre Nierenzellkarzinome zeigen häufig eine Trisomie 7 und/oder 17. Chromophobe Nierenzellkarzinome hingegen weisen Verluste von Chromosomen 1, 2, 6, 10, 13 und 17 auf.5
Eine FISH-Analyse ermöglicht auch die Diagnose von TFE3- und TFEB- Translokationskarzinomen, welche mit konventioneller Histologie alleine nicht sicher von anderen Nierenzellkarzinomen unterscheidbar sind. Die Translokationskarzinome treten vorwiegend bei Kindern und jungen Erwachsenen auf.6 Bei Erwachsenen (> 40 J.) zeigen TFE3-Translokationkarzinome meist eine schlechte Prognose. TFEB-Translokationen sind hingegen mit einem besseren klinischen Verlauf verbunden, eine Ausnahme bilden Karzinome mit einer TFEB-Amplifikation.7
Nierenzellkarzinome können auch hereditär auftreten, bedingt durch pathogene Genvarianten in der Keimbahn. Das mit klarzelligen Nierenzellkarzinomen assoziierte Von-Hippel-Lindau-Syndrom ist durch VHL-inaktivierende Genvarianten bedingt. Aktivierende Mutationen im Protoonkogen MET verursachen papilläre Nierenzellkarzinome, die bilateral und multifokal auftreten können.8 Patienten mit dem Birt-Hogg-Dube-Syndrom weisen eine Folliculin-Genmutation auf und entwickeln chromophobe Nierenzellkarzinome, Onkozytome und Hybridtumoren. Keimbahnmutationen in Succinate-Dehydrogenase-Genen und im Fumarat-Hydratase-Gen (in Verbindung mit hereditärer Leiomyomatose) sind weitere sehr seltene Ursachen für hereditäre Renalzellkarzinome.9, 10
Urothelkarzinom
Die papillären nichtmuskelinvasiven Low-Grade-Urothelkarzinome sind durch aktivierende Mutationen in verschiedenen die Proliferation aktivierenden Onkogenen wie FGFR3, H-RAS oder PIK3CA sowie Deletion des Tumorsuppressorgen CDKN2A gekennzeichnet und weisen insgesamt wenige genetische Veränderungen auf. Die muskelinvasiven, solide wachsenden, High-Grade-Urothelkarzinome hingegen zeichnen sich durch eine sehr hohe genetische Instabilität mit Akkumulation einer Vielzahl von Mutationen aus. Kennzeichnend sind hier sehr früh auftretende TP53-Alterationen, die bereits in In-situ-Vorläuferläsionen mit hochgradiger Dysplasie nachweisbar sind. In einer Analyse von 131 muskelinvasiven Urothelkarzinomen im Rahmen des The Cancer Genome Atlas Project waren 32 Gene rekurrent mutiert, wobei Gene der EGFR-Familie, FGFR3, MAP-Kinase, PI3K-AKT-mTOR sowie den Zellzyklus und das Chromatin regulierende Gene am häufigsten betroffen waren.11, 12 Das muskelinvasive Urothelkarzinom zählt neben Melanom und Lungenkarzinom zu den Tumoren mit der höchsten Mutationslast.13 Obwohl das Urothelkarzinom genetisch gut charakterisiert ist, fehlt noch eine breite Übertragung des Wissens in die klinische Anwendung oder ist noch auf klinische Studien beschränkt. Ein Beispiel für die Translation der Tumorgenetik in die Klinik ist die Entwicklung von Pan-FGFR Inhibitoren wie Erdafitinib zur Behandlung von FGFR3-mutierten Urothelkarzinomen.14
Das Harnblasenkarzinom wird, vor allem basierend auf Genexpressionsstudien, in molekulare Subtypen unterteilt. Eine extensive molekulare Analyse von 415 muskelinvasiven Harnblasenkarzinomen führte zur Differenzierung von 5 molekularen Subtypen13: luminal papillär (35 %), luminal infiltrierend (19 %), luminal (6 %), basal squamös (35 %) und neuronal (5 %). Diese Subtypen korrelieren mit unterschiedlicher Prognose und Ansprechen auf Chemo- und Immuntherapien.13 In der nahen Zukunft werden daher Therapieentscheidungen beim Urothelkarzinom nicht mehr nur auf der Basis von histologischem Tumortyp, Grading und Stadium erfolgen, sondern mit Einbeziehung einer molekularpathologischen Tumorcharakterisierung.
In der zytologischen Harndiagnostik steht schon seit über 10 Jahren ein FISH-Test zur Früherkennung von Urothelkarzinomen bei Risikopatienten und als Überwachung von Patienten mit Rezidivrisiko zur Verfügung. Dieser sogenannte UroVysion®-Test erfasst numerische Aberrationen der Chromosomen 3, 7 und 17 sowie eine Deletion von CDKN2A (9p21).15 Allerdings ist der Einsatz des Tests z. B. wegen eines niedrigen positiv prädiktiven Wertes, der limitierten klinischen Bedeutung von vermehrter Detektion von Low-Grade-Läsionen und der Untersuchungskosten umstritten.15
Das Urothelkarzinom des oberen Harntraktes („upper tract urothelial cancer“/UTUC) ist bei 1–3 % mit einem Lynch-Syndrom assoziiert.16 Es ist nach Kolon- und Endometriumkarzinom die dritthäufigste Neoplasie bei diesem Tumorsyndrom, welches durch Defekte in den DNA-Reparaturproteinen MLH1, MSH2, MSH6 und PMS2 verursacht wird. Zur Identifikation eines Lynch-Syndroms sollten Urothelkarzinome des oberen Harntraktes mittels Immunhistologie auf die Expression der vier DNA-Reparaturproteine untersucht werden. Bei Verlust der Expression eines der Reparaturproteine in den Karzinomzellen muss zur weiteren Abklärung eine molekulare Mikrosatellitenanalyse erfolgen, weil bei UTUC ein Verlust der Proteinexpression bei einem signifikanten Anteil der Patienten nicht mit der für das Lynch-Syndrom typischen hohen Mikrosatelliteninstabilität verbunden ist.16–18 Das Patientenalter ist jedenfalls kein Stratifizierungsfaktor für die Testung, weil die Lynch-Syndrom-assozierten UTUC erst in höherem Lebensalter auftreten (mittleres Alter 69 Jahre).16
Prostatakarzinom
Molekularpathologische Untersuchungen haben derzeit weder für die Diagnostik noch für die Behandlung des Prostatakarzinoms einen großen Stellenwert. Vor allem das Auffinden von Biomarkern als prognostische Parameter für die Einschätzung des Tumorverhaltens ist jedoch von großem Interesse.19 Unter einer Vielzahl von potenziellen Biomarkern ist die Inaktivierung von PTEN durch Deletion, Promotermethylierung oder Mutation als Marker zur Unterscheidung von indolenten und aggressiven Tumoren vielversprechend.20 Der Verlust einer PTEN-Expression wird mittels Immunhistochemie nachgewiesen, Mutationen und Deletionen werden mittels NGS und FISH erfasst.
Im Kontext der Zweitlinientherapie beim kastrationsresistenten metastasierten Prostatakarzinom könnte der Nachweis einer Androgenrezeptor-Splicevariante-7 (AR-V7) die Entscheidung zwischen einer Therapie mit Androgen-Rezeptor-Inhibitoren wie Abirateron und Enzalutamid und einer Chemotherapie mit Taxanen unterstützen.21 Das AR-V7-Molekül behält die DNA-Bindungsdomäne, verliert aber die regulatorische Ligandenbindungsdomäne und ist dadurch konstitutiv aktiviert. Tumoren mit AR-V7-Expression sind resistent gegenüber Abirateron oder Enzalutamid, können aber Sensitivität gegenüber Taxanen aufweisen.
Hodentumoren
Bei Teratomen oder auch in seltenen Fällen bei Rezidiven kurativ behandelter Keimzelltumoren kann es zum Auftreten einer somatischen Malignität (vormals somatische Transformation) kommen. In diesen Fällen ist eine rein histomorphologische Unterscheidung des Tumorgewebes einer somatischen Malignität von primären Neoplasien nicht immer möglich. Eine Zuordnung zu einem Keimzelltumor kann durch den Nachweis des Isochromosoms 12p mittels FISH-Analyse erfolgen, da es bei der Entwicklung von einer In-situ-Keimzellneoplasie zu einem (malignen) Keimzelltumor unter anderem auch zu einem Zugewinn des kurzen Arms des Chromosoms 12, vorwiegend als Isochromosom, kommt.22, 23
Zusammenfassend sind urologische Tumoren umfassend genetisch charakterisiert, und viele potenzielle Biomarker werden in Studien evaluiert. Derzeit sind genetische Biomarker in der molekularen Pathologie allerdings vorwiegend als Unterstützung der histopathologischen Diagnostik in Verwendung. In Einzelfällen werden aber schon jetzt bei auf Standardtherapien refraktären urologischen Tumoren ausgedehntere molekulare Profile mittels „next-generation sequencing“ und erweiterter Immunhistologie erstellt, um Zielmoleküle für eine experimentelle Therapie zu identifizieren.
1 Müllauer L, Memo 2017; 10(4):244–247
2 Müllauer L, Memo 2017; 10(1):42–45
3 Stuhler V et al., World J Urol 2018; Oct. 29 [Epub ahead of print]
4 Posadas EM et al., Nature Reviews Nephrology 2017; 13(8):496–511
5 Inamura K, Int J Mol Sci 2017; 18(10):pii:E2195
6 Rupp NJ et al., European Urology Supplements 2017; 16(12):241–252
7 Argani P et al., Am J Surg Pathol 2016; 40(11):1484–1495
8 Schmidt L et al., Nat Genet 1997; 16(1):68–73
9 Ricketts CJ et al., J Urol 2012; 188(6):2063–2071
10 Grubb RL et al., J Urol 2007; 177(6):2074–2079; discussion 2079–2080
11 Weinstein JN et al., Nature 2014; 507(7492):315–322
12 Knowles MA, Hurst CD, Nature Reviews Cancer 2014; 15(1):25–41
13 Robertson AG et al., Cell 2017; 171(3):540–556.e25
14 Hanna KS, J Oncol Pharm Pract 2018 Oct. 10; 1078155218805141
15 Bubendorf L, Acta Cytologica 2011; 55(2):113–119
16 Ju JY et al., Am J Surg Pathol 2018; 42(11):1549–1555
17 Metcalfe MJ et al., J Urol 2018; 199(1):60–65
18 Olagui GS et al., Bull Cancer 2014; 101(2):144–150
19 Kretschmer A, Tilki D, Crit Rev Oncol Hematol 2017; 120(Dec.):180–193
20 Jamaspishvili T et al., Clinical implications of PTEN loss in prostate cancer. Nat Rev Urol 2018; 15(4):222–234
21 Scher HI et al., JAMA Oncol 2018; 4(9):1179–1186
22 Lutke Holzik MF et al., Ann Surg Oncol 2003; 10(2):131–135
23 Verdorfer I, Pathologe 2014; 35(3):218–223

AutorIn: Dr. Felicitas Oberndorfer
Klinisches Institut für Pathologie, Medizinische Universität Wien/AKH Wien

AutorIn: Ao. Univ.-Prof. DDr. Leonhard Müllauer
Klinisches Institut für Pathologie, Medizinische Universität Wien/AKH Wien

Ursprünglich erschienen:
SU 04|2018
SU 04|2018