


























































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Familiäre Hypercholesterinämie im Kindesalter
Früh erkennen, Leben retten
30. April 2026
Die familiäre Hypercholesterinämie (FH) wird autosomal-dominant vererbt und zählt zu den häufigsten genetischen Stoffwechselerkrankungen. Die Prävalenz beträgt heterozygot etwa 1: 250, homozygot etwa 1:250.000. Bei heterozygoten Betroffenen liegt die LDL-Konzentration zwischen 190 und 400 mg/dl, bei homozygoten können Werte von 500 bis 1.000 mg/dl erreicht werden. Das Vererbungsmuster führt dazu, dass in jeder Generation Familienmitglieder betroffen sein können.
Historische Entwicklung der Forschung
Die Erkrankung wurde bereits im 19. Jahrhundert als gelbliche Haut- und Sehnenveränderungen (Xanthome, Abb.) mit schweren Gefäßveränderungen beschrieben. Einen wichtigen wissenschaftlichen Beitrag lieferte Carl Müller, der 1939 den Zusammenhang zwischen hereditärer Xanthomatose und Angina Pectoris beschrieb und damit die genetische Grundlage der Erkrankung hervorhob.
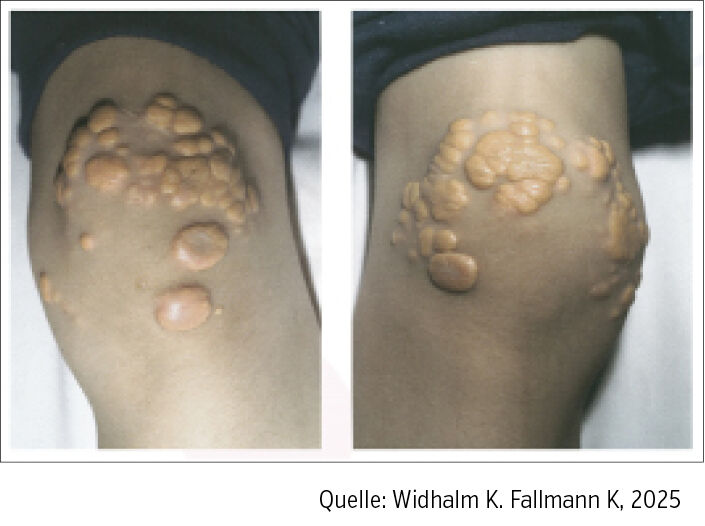
Abb.: Xanthome eines 12-jährigen Buben mit homozygoter FH
Diagnose der FH
Die Diagnose erfolgt klinisch (Familienanamnese, frühzeitige kardiovaskuläre Ereignisse, erhöhte LDL-Werte von 130–190 mg/dl) und/oder genetisch. Ein positiver Gentest bestätigt die Erkrankung, ein negatives Ergebnis schließt sie jedoch nicht aus. Bei Kindern mit heterozygoter FH zeigen sich früh Gefäßveränderungen wie etwa eine erhöhte Intima-Media-Dicke, was den Beginn der Gefäßveränderungen lange vor dem Auftreten von Symptomen belegt.
Lebensstilmaßnahmen
Vor Beginn einer medikamentösen Therapie sind Lebensstilmaßnahmen die entscheidende Säule. Eine gesunde, fettbewusste Ernährung mit wenig gesättigten Fettsäuren (max. 10 %), ausreichend Ballaststoffen und wenig Zucker und Fertigprodukten sowie regelmäßige körperliche Aktivität senken das LDL-Cholesterin und verbessern das kardiovaskuläre Risiko. Besonders bei Kindern sollten diese Maßnahmen früh gemeinsam mit der Familie etabliert werden, sie bleiben auch unter Medikation zentral für das langfristige Management.
Medikamentöse Therapie
Die medikamentöse Therapie der FH zielt auf eine deutliche LDL-Senkung ab. Statine sind Standardtherapie und auch bei Kindern wirksam und sicher, senken LDL-Werte signifikant und verlangsamen Gefäßveränderungen. Langzeitbeobachtungen zeigen, dass früh begonnene Therapien den LDL-Spiegel dauerhaft stark reduzieren. Neuerdings stehen PCSK9-Inhibitoren, Lomitapid und RNA-basierte Therapien zur Verfügung. Evolocumab konnte in einer Studie bei Jugendlichen LDL um 44 % senken.
Fazit
Trotz der klaren diagnostischen Kriterien und effektiven Therapiemöglichkeiten bleibt die FH häufig unerkannt. Ursachen sind unter anderem das Fehlen sichtbarer Symptome im Kindesalter, verbunden mit Verunsicherung und Angst der Eltern, ein begrenztes Bewusstsein für genetische Ursachen von FH sowie Wissenslücken im medizinischen Alltag. Studien zeigen, dass selbst unter Pädiater:innen teilweise Unsicherheiten hinsichtlich Prävalenz, Risikoabschätzung und Therapie bestehen.
Die FH ist eine gut charakterisierte, aber weiterhin häufig übersehene genetische Stoffwechselerkrankung. Eine frühzeitige Diagnose im Kindesalter, verbunden mit adäquater Therapie, kann das Risiko schwerer kardiovaskulärer Ereignisse deutlich reduzieren.
Praxismemo
- Die FH zählt zu den häufigsten genetischen Stoffwechselerkrankungen, wird aber oft übersehen.
- Die Diagnose erfolgt anhand Familienanamnese, erhöhter LDL-Werte (> 130 mg/dl) und/oder genetischer Testung.
- Lebensstilmaßnahmen (Ernährung, regelmäßige körperliche Aktivität) sind die Basis jeder Therapie.
- Eine früh begonnene medikamentöse Therapie reduziert nachhaltig das kardiovaskuläre Risiko.
- Statine senken das LDL-Cholesterin deutlich und sind auch bei Kindern wirksam und sicher.

Autor:
Univ.-Prof. Dr Kurt Widhalm
Univ.-Prof. Dr Kurt Widhalm
Österreichisches Akademisches Institut für Ernährungsmedizin, 1090 Wien

Autorin:
Mag.a Karin Fallmann
Mag.a Karin Fallmann
Österreichisches Akademisches Institut für Ernährungsmedizin, Wien

Ursprünglich erschienen:
AEK 09|2026
AEK 09|2026
KLINISCHE RELEVANZ UND FRÜHE MANIFESTATIONEN
Die klinische Symptomatik zeigt sich meist erst im Erwachsenenalter. Eine frühzeitige Identifikation und Therapie kann die Entwicklung der Atherosklerose hintanhalten. Bei homozygoter familiärer Hypercholesterinämie können bereits Kinder Koronarverschlüsse und lebensbedrohliche Komplikationen entwickeln.
RED FLAGS
LDL-Cholesterin >130–160 mg/dl bei Kindern bzw. erhöhte Werte bei positiver Familienanamnese sollten an eine familiäre Hypercholesterinämie denken lassen. Weitere Hinweise sind frühzeitige kardiovaskuläre Ereignisse bei Verwandten 1. Grades sowie Xanthome. Frühzeitiges Handeln ist entscheidend.
LANGZEITRISIKO
Nicht nur der aktuelle LDL-Wert, sondern vor allem die Dauer der Exposition bestimmt das kardiovaskuläre Risiko. Eine früh beginnende LDL-Erhöhung führt über die Jahre zu einer höheren Cholesterol Burden und beschleunigt die Atherogenese. Eine rechtzeitig initiierte Therapie kann dieses kumulative Risiko deutlich reduzieren.
TIPPS FÜR DIE PRAXIS
- Bei erhöhtem LDL immer Familienanamnese erheben und bei Verdacht Angehörige im Rahmen eines Kaskaden-Screenings untersuchen.
- Bei familiärem Risiko Lipidwerte früh – möglichst vor dem 10. Lebensjahr – bestimmen und regelmäßig kontrollieren.
- Ernährung und Bewegung zuerst: Fettbewusste Kost und körperliche Aktivität senken LDL und sind Basis jeder FH-Therapie.
- Eine medikamentöse Therapie ist wirksam und wird meist sehr gut vertragen.
- Erkrankung und Therapie den Betroffenen verständlich erklären– gute wissenschaftlich fundierte Information stärkt die Prävention und Compliance.
WAS PATIENT:INNEN WISSEN WOLLEN
Kann FH nur durch Ernährung und Bewegung behandelt werden?
Eine gesunde Ernährung und ausreichend körperliche Bewegung sind die Basis der Therapie, reichen meist aber nicht aus. Da die Ursache ein genetischer Defekt ist, sind häufig Medikamente notwendig, um die LDL-Cholesterinwerte ausreichend zu senken.
Ist eine medikamentöse Behandlung bei Kindern wirklich notwendig und sicher?
Bei deutlich erhöhten LDL-Werten ist eine medikamentöse Therapie unumgänglich. Studien zeigen, dass Statine bei Kindern wirksam und in der Regel gut verträglich sind, um das spätere Risiko für Herz-Kreislauf-Erkrankungen senken können.
Quellenverzeichnis:
Zusammenfassung des Vortrags von Univ.-Prof. Dr. Kurt Widhalm im Zuge des Festsymposiums des Österreichischen Akademischen Instituts für Ernährungsmedizin zum Thema „Familiäre Hypercholesterinämie – Prävention beginnt in der Familie“ vom 20.2.2026
Bildnachweis
Vorschaubild: © Widhalm K. Fallmann K, 2025