


























































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Psychische Störungen bei Chorea Huntington
22. Juni 2011
Chorea Huntington ist eine autosomal-dominant vererbte Erkrankung mit einer Prävalenz von 6 bis 12 Betroffenen/100.000 Personen in West – europa und Nordamerika. In Österreich wird die Zahl der an Chorea Huntington Erkrankten auf 500 bis 1.000 geschätzt. Die Chorea Huntington gehört damit – trotz der niedrigen Prävalenz – zu den häufigsten genetisch bedingten neurologisch-psychiatrischen Erkrankungen. Die Ursache der Chorea Huntington ist eine Genmutation auf dem kurzen Arm des Chromosom vier. Bei der Mutation handelt es sich um eine Verlängerung der Wiederholungen des Basen-Tripletts CAG1. Die Expansion des Gens führt zur Bildung von langen Polyglutaminen. Durch den Einbau der Polyglutamine in das Protein Huntington erfolgt eine Stukturumwandlung des Proteins, die vor allem im Gehirn eine Apoptose einleitet. Die Apoptose häuft sich in bestimmten Hirnregionen, vor allem im Striatum und dort speziell im Nucleus caudatus. Im fortgeschrittenen Stadium weist das Gehirn eine deutliche globale Atrophie auf, bedingt vor allem durch eine kortikale Atrophie und eine Atrophie der benachbarten weißen Substanz2. Bei den meisten Betroffenen liegt der Krankheitsbeginn im mittleren Erwachsenenalter, zwischen dem 35. und 45. Lebensjahr.
Klinische Charakteristika
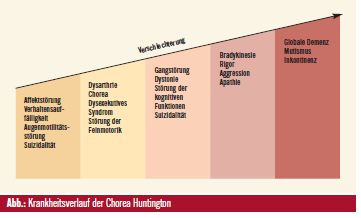
Bereits George Huntington, der Namensgeber der Erkrankung, beschrieb 1872 in Ohio psychische Symptome als typische Kennzeichen der Erkrankung: “Die Krankheit ist erblich, es gibt einer Neigung zur Verrücktheit und zum Suizid, schwere Symptome treten nur im Erwachsenenalter auf.”3 Mit dieser Definition grenzte George Huntington die Chorea Huntington von der Chorea minor ab, der Folge einer Streptokokkeninfektion. Heute wird die typische Klinik der Chorea Huntington als eine Symptomtrias aus neurologischen Ausfällen, psychiatrischen Symptomen und einer subkortikalen Demenz beschrieben. Neurologische Symptome wie choreatische Bewegungen der Extremitäten, des Kopfes sowie des Rumpfes, Sprech- und Schluckstörungen, Augenmotilitätsstörungen, im späten Stadium Gangstörungen und Störungen der Stellreflexe werden häufig als die typischen, wichtigen und behandlungsbedürftigen Symptome der Erkrankung angesehen. Im Gegensatz dazu werden psychische und psychiatrische Symptome oft als “Nebenschauplatz” abgehandelt. So werden psychiatrische Symptome der Chorea Huntington in Lehrbüchern meist gar nicht angeführt. Dabei stellt sich die Realität in der täglichen Praxis und im Umgang mit den Patienten anders dar: Bei einer Befragung der Patienten und der Angehörigen zeigt sich, dass psychiatrische Probleme der Patienten viel weitreichendere Konsequenzen haben als motorische Ausfälle. So sind psychische Probleme oft der limitierende Faktor, wegen dem Patienten nicht mehr von Angehörigen zu Hause betreut werden können, sondern im Pflegeheim untergebracht werden müssen. Angehörige und Patienten leiden meist auch weniger unter den körperlichen Beeinträchtigungen der Patienten als unter einer schlechten psychischen Verfassung. Auch wenn psychische Symptome ein prominenter Teil der Erkrankung sind, so wird klassischerweise der Beginn der Chorea Huntington mit dem ersten Auftreten von neurologischen Symptomen definiert, auch wenn psychische Symptome den neurologischen Ausfällen typischerweise bereits um Jahre vorausgehen. Psychische Symptome der Chorea Huntington verändern sich stark im Laufe der Erkrankung und lassen sich in zwei Gruppen unterteilen: einerseits die psychischen Symptome präsymptomatischer Patienten und von Patienten in frühen Stadien und andererseits psychische Symptome später Stadien.
Frühe psychische Symptome
Gerade affektive Erkrankungen sind bei Patienten bereits vor dem Auftreten von neurologischen Symptomen und somit vor dem offiziellen Beginn der Erkrankung häufig. So haben präsymptomatische Patienten ein signifikant höheres Risiko, an einer unipolaren Depression zu erkranken, als Personen ohne die genetische Veranlagung für Chorea Huntington4. Bereits Jahre vor dem Auftreten erster neurologischer Symptome erfüllten in einer Studie 15% der präsymptomatischen Patienten die DSM-IV-Diagnose der Major Depression. Die Wahrscheinlichkeit, an einer Depression zu erkranken, nimmt zu, je näher diese Personen dem Zeitpunkt des Auftretens der ersten neurologischen Symptome – und somit dem offiziellen Ausbruch der Erkrankung – kommen. Während sich 10 Jahre vor Krankheitsbeginn die Prävalenz der Depression bei präsymptomatischen Personen nicht von der der Allgemeinbevölkerung unterscheidet, steigt 1 bis 5 Jahre vor Krankheitsausbruch die Häufigkeit einer Depression bei präsymptomatischen Personen auf 20% an.
Zur Genese der Depression in frühen Stadien wurde lange vermutet, dass die starke psychische Belastung durch die neue Diagnose und der ständige Ausnahmezustand vor allem am Anfang der Erkrankung zu einem erhöhten Stresslevel und damit zu einem vermehrten Auftreten von Depressionen führt. Mittlerweile zeigen jedoch mehrere Untersuchungen, dass auch Patienten, die nichts von ihrer Veranlagung für Chorea Huntington wissen, signifikant häufiger an affektiven Erkrankungen erkranken als Personen, die negativ für das Huntington-Gen sind, aber auch in Familien mit Chorea Huntington leben und somit den gleichen Belastungen ausgesetzt sind4. So ist es wahrscheinlich, dass nicht nur die exogene Belastung durch die Familien und die ständige Beschäftigung mit Chorea Huntington die Depression auslösen, sondern die bereits stattgehabte organische Veränderung im Gehirn und es sich – zumindest teilweise – um eine organisch bedingte Depression handelt.
Einige Untersuchungen zeigen bei präsymptomatischen Patienten ein erhöhtes Auftreten von Zwangshandlungen und Zwangsgedanken, wobei hier Zwangsgedanken überwiegen5, 6. Allerdings tritt der Zwang angesichts der Schwere und Häufigkeit affektiver Erkrankungen deutlich in den Hintergrund. Angsterkrankungen und Psychosen scheinen in frühen Phasen etwas häufiger aufzutreten als in der Allgemeinbevölkerung, wobei sich hier die Ergebnisse der Studien stark von einander unterscheiden5. Andere psychiatrische Erkrankungen wie Manie oder Persönlichkeitsstörungen kommen in diesem frühen Stadium nicht häufiger vor als in der Allgemeinbevölkerung7. Während es in späteren Stadien zu einer deutlichen Veränderung des klinischen Bildes der psychischen Störungen kommt, unterscheidet sich die psychische Situation von präsymptomatischen Patienten und Patienten in frühen symptomatischen Stadien nicht. Offensichtlich sind psychiatrische Defizite vor allem am Anfang der Erkrankung stabil und ändern sich erst im langfristigen Verlauf der Erkrankung.
Späte psychische Symptome
Auch wenn ein Teil der Patienten im fortgeschrittenen Stadium weiterhin von affektiven Störungen, Zwangs- und Angststörungen begleitet wird, kommt es hier doch meist zu einer deutlichen Veränderung des psychischen Zustandsbilds. In diesem Stadium stehen frontale Verhaltensmuster mit Reizbarkeit, Aggression oder Apathie im Vordergrund. Dies erklärt sich aus der engen Verbindung zwischen Basalganglien und frontalem Kortex, die bei diesen Patienten zum bekannten Bild des “Frontalhirnsyndroms” führt. Zusätzlich finden sich bei den meisten Patienten, zumindest in manchen Phasen, Affektlabilität, Euphorie, Gleichgültigkeit, Beeinträchtigung der Steuerungsfähigkeit, Haltlosigkeit, Affektinkontinenz oder Impulshandlungen.
Aggression ist mit einer Prävalenz von 60% in späten Stadien eine der am häufigsten auftretenden psychischen Störungen bei Chorea Huntington8. Gerade in Pflegeheimen ist die körperliche und verbale Aggression ein schwieriges Thema: Ein Drittel aller Pflegeheimbewohner zeigten in einer Untersuchung ein schweres und häufiges aggressives Verhalten9. Am häufigsten waren dabei Pflegepersonen verbaler Aggression ausgesetzt, gefolgt von körperlichen Angriffen auf die Pflegepersonen. Seltener kamen körperliche Aggressionen gegen Mitpatienten oder gegen den Patienten selbst vor.
Anders als fortgeschrittene neurologische Symptome wie Gang- oder Schluckstörungen sind psychische Symptome wie Aggressivität und Reizbarkeit oft für viele Angehörige der ausschlaggebende Grund, Patienten nicht mehr zu Hause versorgen zu können oder zu wollen. Da diese späten psychischen Störungen wegen der fortgeschrittenen zerebralen Atrophie auch nicht immer optimal auf eine psychopharmakologische Behandlung ansprechen, kann oft nur eine geringe Reduktion der Belastung erreicht werden. Eine Befragung der Patienten ergab, dass sich die meisten Personen selbst ihrer Reizbarkeit und Aggression bewusst sind, sie aber selbst schwer modifizieren können. Patienten beschreiben, dass sie durch viele gleichzeitige externe Stimulationen überfordert sind und deshalb nicht adäquat reagieren können. Während auch bei manchen Patienten schon in frühen Stadien apathische Zustände auftreten können, verstärken sich diese im Verlauf der Erkrankung meist deutlich. Apathie wurde bei einer Untersuchung unter Huntingtonpatienten bei 44% aller Patienten gefunden, wobei bei diesen Patienten Depressionen ausgeschlossen wurden10. Gerade in späten Stadien, wenn Patienten mit Chorea Huntington schlechter kommunizieren können, wird Apathie oft mit Depression verwechselt.
Angehörige benötigen Aufklärung darüber, dass Anzeichen wie Interessenlosigkeit oder mangelnde Eigeninitiative auf einen apathischen Zustand hinweisen können und nicht depressive Anzeichen sein müssen. Angehörige sollten geschult werden, den Patienten positiv zu motivieren, ihn zu fördern, aber auch nicht zu unterfordern, ihn nicht unter Druck zu setzen und ihm sein eigenes Tempo zu lassen. Die für Personen im Umkreis des Patienten belastenden psychischen Spätsymptome können sich im Laufe der Erkrankung rasch verändern. Angehörige erzählen, dass die wechselnden Phasen der frontalen Enthemmung/ Apathie schnell wechseln und durch externe Stimuli wie beruhigende Worte oder ein beruhigendes Umfeld schwer veränderbar sind.
Suizidalität
Da gerade in frühen Stadien der Chorea Huntington die Depressionsrate stark erhöht ist, muss bei diesen Patienten natürlich ein besonderes Augenmerk auf Suizidalität und Suizidversuche gelegt werden. Tatsächlich ist die Suizidrate bei Patienten mit Chorea Huntington deutlich erhöht: Man geht von bis zu 13% erfolgreichen Suiziden bei Patienten mit Chorea Huntington aus; dies ist eine massive Erhöhung im Vergleich zur Allgemeinbevölkerung mit 15 Suiziden/100.000 Personen pro Jahr. Suizidversuche werden von bis zu einem Viertel aller Patienten mit Chorea Huntington unternommen11. An Suizid denken oder über Suizidgedanken berichten zumindest die Hälfte der betroffenen Personen. Die Risikofaktoren für Suizid bei Huntingtonpatienten unterscheiden sich nicht von denen der Allgemeinbevölkerung: Hier zählen ebenfalls depressive Personen, Kinderlose, Alleinstehende und Personen mit funktionellen Einschränkungen zur Risikogruppe.
Doch im Gegensatz zur weitverbreiteten Meinung ist nicht die Zeit der genetischen Testung und der Aufklärung über den positiven Gentest einer der vulnerabelsten Punkte. Tatsächlich kann die Tatsache, endlich eine Diagnose erhalten und Gewissheit über den weiteren Verlauf der Erkrankung zu haben, die Suizidgefahr reduzieren.
Es lassen sich zwei andere kritische Zeitpunkte mit einer deutlich erhöhten Suizidgefahr identifizieren12: Zum einen der Zeitpunkt des Auftretens der ersten neurologischen Symptome. Hier bemerken Patienten das erste Mal eigene Defizite, die sie nicht mehr kompensieren können und müssen sich der Tatsache stellen, nun wirklich an einer unheilbaren Krankheit erkrankt zu sein. Oft fühlen sie sich an das Schicksal von nahen Familienangehörigen erinnert, die in kurzer Zeit physisch und psychisch schwer beeinträchtigt wurden. Der zweite kritische Zeitpunkt ist das mittlere Krankheits – stadium. In diesem Stadium verlieren Patienten oft ihre autonomen Fähigkeiten (Verlust des Arbeitsplatz, Einschränkungen beim Autofahren, Verlust der finanziellen Unabhängigkeit etc.) und werden zunehmend von der Umwelt abhängig. Die meisten dieser Funktionen werden nicht freiwillig aufgegeben, was zu einer ungewollten Einschränkung führt. In beiden vulnerablen Stadien sind die Patienten aber durchaus kognitiv und motorisch noch gut in der Lage einen Suizid zu planen und durchzuführen, während die Abhängigkeit von der Umwelt und die Angst vor der Erkrankung wachsen.
resümeeDie kombinierte neurologische und psychiatrische Betreuung von Patienten mit Chorea Huntington ist international mittlerweile Standard. Da psychiatrische und psychische Probleme in jedem Stadium der Erkrankung zu einem hohen Prozentsatz auftreten, ist die ständige Begleitung des Patienten, am besten bereits vor dem Auftreten von ersten neurologischen Symptomen, unbedingt zu empfehlen. Denn die größten Probleme für Angehörige, Pflegepersonen und den Patienten selbst erstehen meist nicht durch neurologische Symptome, sondern durch typische psychiatrische Erkrankungen wie Depressionen oder Aggressionen.
1) Huntington’s Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. 1993; 72(6):971-983
2) Tellez-Nagler I, Johnson A, Terry R, Ultrastructural and histochemical study of cerebral cortex biopsies in Huntington’s disease. Advances in Neurology. 1973(1):397-398
3) Huntington G. On chorea, The Medical and Surgical Reporter 1872
4) Julien CL, Thompson JC, Wild S et al., Psychiatric disorders in preclinical Huntington’s disease. J Neurol Neurosurg Psychiatry. 2007;78(9):939-943
5) van Duijn E, Kingma EM, van der Mast RC, Psychopathology in verified Huntington’s disease gene carriers. J Neuropsychiatry Clin Neurosci. 2007; 19(4):441-448
6) Craufurd D, Thompson JC, Snowden JS, Behavioral changes in Huntington Disease. Neuropsychiatry Neuropsychol Behav Neurol. 2001; 14(4):219-226
7) van Duijn E, Kingma EM, Timman R et al., Cross-sectional study on prevalences of psychiatric disorders in mutation carriers of Huntington’s disease compared with mutation-negative first-degree relatives. J Clin Psychiatry. 2008; 69(11):1804-1810
8) Marder K, Zhao H, Myers RH et al., Rate of functional decline in Huntington’s disease. Huntington Study Group. Neurology. 25 2000; 54(2):452-458
9) Nance MA, Sanders G, Characteristics of individuals with Huntington disease in long-term care. Mov Disord. 1996; 11(5):542-548
10) Caine ED, Shoulson I, Psychiatric syndromes in Huntington’s disease. Am J Psychiatry. 1983; 140(6):728-733
11) Anderson KE, Marder KS, An overview of psychiatric symptoms in Huntington’s disease. Curr Psychiatry Rep. 2001; 3(5):379-388
12) Paulsen JS, Hoth KF, Nehl C, Stierman L, Critical periods of suicide risk in Huntington’s disease. Am J Psychiatry. 2005; 162(4):725-731

Ursprünglich erschienen:
SP 02|2011
SP 02|2011