


























































































Neu Registrieren
Ich habe noch kein Benutzerkonto und möchte mich kostenlos registrieren.
Zur Registrierung
Mechanismen des erhöhten Diabetesrisikos unter Statintherapie
4. März 2013

Der Nutzen der Lipidsenkung mit Statinen in Hinblick auf die kardiovaskuläre Risikosenkung steht bei Diabetikern ebenso wie bei Nichtdiabetikern außer Streit (CTT Collaborators, Lancet 2005; Lancet 2008; Lancet 2012). Allerdings hat sich die mit der HMG-CoA-Reduktasehemmung offenbar assoziierte Erhöhung des Diabetesrisikos in den letzten Jahren als relevante Schattenseite der Therapie herausgestellt. Vermehrte Diabetesmanifestationen traten vor allem in Studien zu Tage, in denen sehr niedrige Serumspiegel an LDL-Cholesterin (LDL-C) erreicht wurden (Preiss et al., JAMA 2011). Der diabetogene Effekt der Statine ist durch insgesamt drei Metaanalysen (Sattar et al., Lancet 2010; Preiss et al., JAMA 2011, Abb. 1; Waters et al., J Am Coll Cardiol 2013) zweifelsfrei erwiesen.
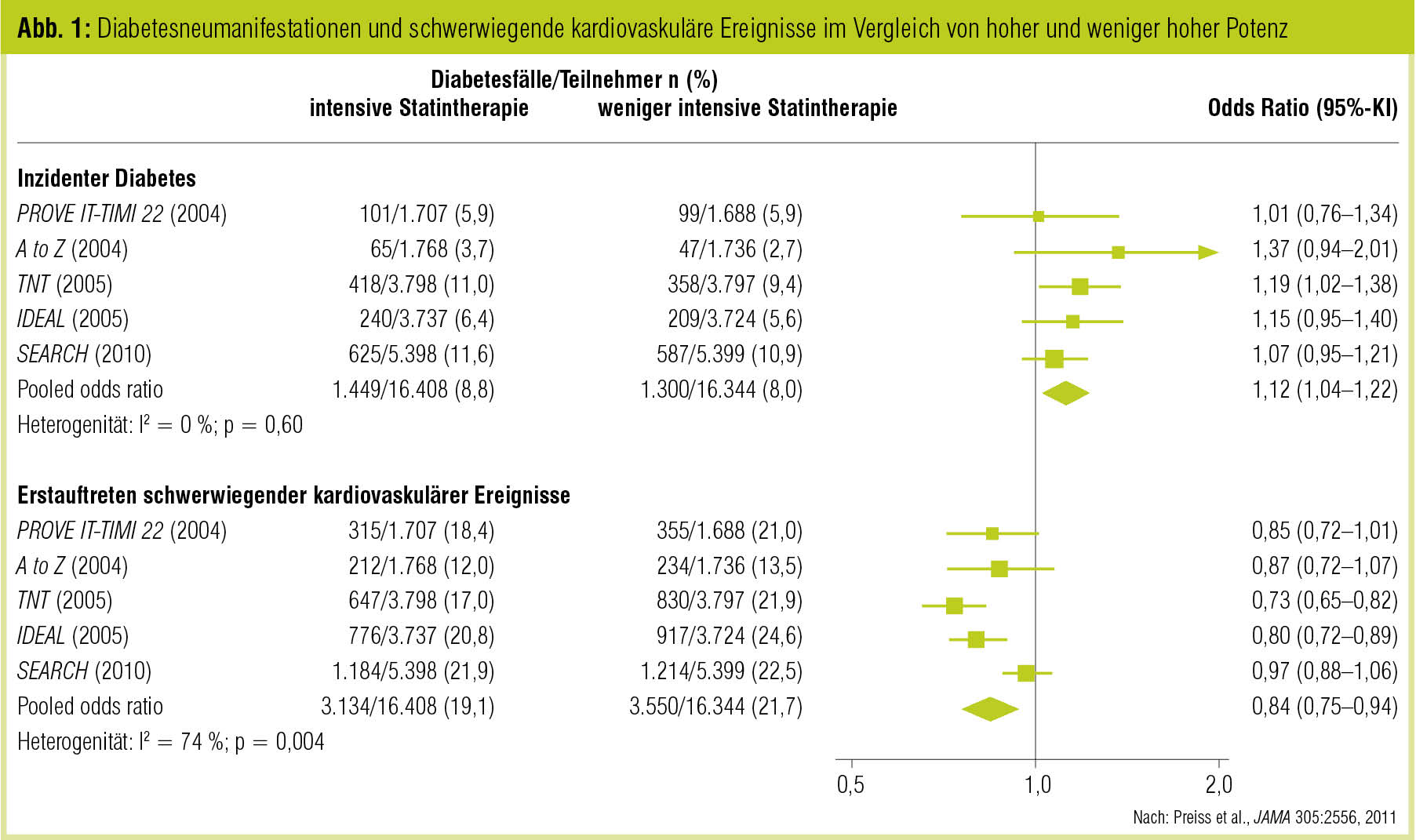
In Expertenkreisen herrscht dennoch die Meinung vor, dass das erhöhte Risiko für die Entwicklung eines Typ-2-Diabetes unter Statintherapie keinen Grund darstellt, auf diese Therapieoption zu verzichten, da der kardiovaskuläre Nutzen das Diabetesrisiko überwiegt. Diese Einschätzung dürfte sich aber ändern, sobald lipidsenkende Medikamente ohne diabetogenes Potenzial zur Verfügung stehen; derzeit gibt es eine solche therapeutische Alternative allerdings nicht.
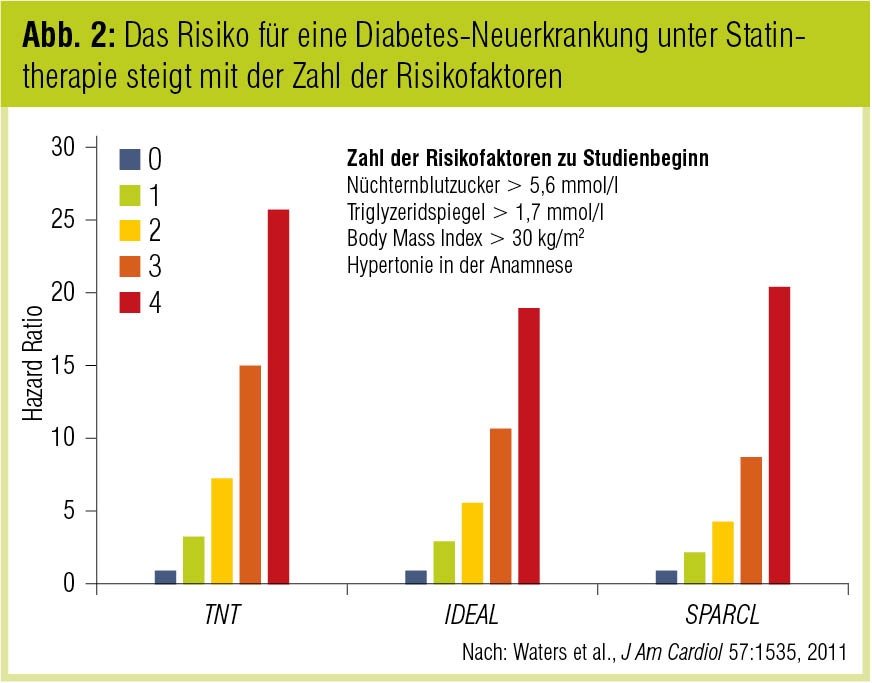
Nicht alle Patienten mit einer therapiebedürftigen Lipidstoffwechselstörung haben unter einer Statintherapie dasselbe Diabetesrisiko. Post-hoc-Analysen der TNT-Studie, der IDEAL-Studie und der SPARCL-Studie zeigen übereinstimmend, dass das Risiko mit der Zahl kardiometabolischer Risikofaktoren – erhöhte Nüchternglukose, erhöhte Triglyzeride, erhöhter Body Mass Index (BMI), Hypertonie in der Anamnese – ansteigt (Waters et al., J Am Coll Cardiol 2013; Abb. 2).
Unveränderte Insulinsensitivität, verringerte Insulinproduktion
Typ-2-Diabetes entwickelt sich prinzipiell auf dem Boden einer Insulinresistenz, die initial durch vermehrte Freisetzung von Insulin kompensiert wird. Zur Diabetesmanifestation kommt es, wenn die Betazellen nicht mehr in der Lage sind, ausreichend Insulin zu produzieren, um die Insulinresistenz zu überwinden. Von Substanzen, welche die Diabetesentstehung begünstigen, ist zu erwarten, dass sie die Insulinfreisetzung und eventuell auch die Insulinsensitivität beeinflussen. Ein alleiniger Effekt auf die Insulinsensitivität reicht für die Diabetesmanifestation nicht aus.
Eine Metaanalyse von 16 klinischen Studien mit insgesamt 1.146 Patienten zeigt, dass Statine keinen signifikanten Effekt auf die Insulinsensitivität haben (Baker et al., Diabetes Res Clin Pract 2010). Dem gegenüber weisen immer mehr Untersuchungen darauf hin, dass Statine das Betazellversagen propagieren. So wurde gezeigt, dass Simvastatin und Atorvastatin die Insulinsekretion aus einer Betazelllinie (MIN6) verringern (Ishikawa et al., J Atheroskler Thromb 2006), für Lovastatin wurde ein solcher Effekt an Ratteninselzellen nachgewiesen (Tsuchiya et al., Endocrinology 2010).
Beeinflussung der Betazellfunktion durch Cholesterin
An isolierten humanen und tierischen Inselzellen konnte demonstriert werden, dass LDL-C bereits in physiologischen Konzentrationen die glukoseabhängige Insulinsekretion (GSIS) hemmt. Der LDL-Rezeptor (LDLR) dürfte an diesem Effekt wesentlich beteiligt sein (Rütti et al., Endocrinology 2009). Kruit et al. (Diabetologia 2010) fanden bei LDLR-Knock-out-Mäusen mit diätetisch induzierter Hypercholesterinämie einen erhöhten Cholesteringehalt der Betazellen und damit assoziiert eine Abnahme der maximalen GSIS sowie überhöhte Blutzuckerwerte im Vergleich zu Wildtyp-Mäusen.
Die Bedeutung des LDLR wird weiters durch die Beobachtung gestützt, dass Patienten mit heterozygoter familiärer Hypercholesterinämie (gekennzeichnet durch einen genetischen Defekt des LDLR) ein vergleichsweise geringes Diabetesrisiko aufweisen (Vohl et al., Eur J Clin Invest 1997; Skoumas et al., Int J Cardiol 2007). Statine könnten sich somit über die LDLR-vermittelte vermehrte Aufnahme von LDL-C in die Betazellen negativ auf die glukoseabhängige Insulinfreisetzung auswirken.
Konsequenzen der HMG-CoA-Reduktasehemmung
Eine weitere Hypothese beruht darauf, dass Statine durch HMG-CoA-Reduktasehemmung die Synthese von Cholesterinvorläufern in der Betazelle hemmen. Ein wichtiges Nebenprodukt der Cholesterinsynthese ist das für die ATP-Produktion notwendige Ubiquinon. Statine könnten über diesen Weg die Verfügbarkeit von ATP und damit die Freisetzung von Insulin aus insulinhaltigen Vesikeln limitieren (Abb. 3). Diese – zellbiologisch plausible – Interferenz zwischen Statintherapie und reduzierter Insulinsekretion ist wissenschaftlich noch nicht belegt. Bewiesen ist aber, dass Cholesterin bzw. Cholesterinvorläufer die Insulinsekretion fördern und dass in die Betazelle aufgenommene Statine die Insulinsekretion hemmen. Der inhibierende Effekt der Statine konnte durch Metabolite der Cholesterinsynthese wie Mevalonat oder Squalen aufgehoben werden. Mikroskopisch lässt sich in Inselzellen unter Statintherapie eine Verschiebung in Richtung weniger großer Granula, die kein Insulin sezernieren, nachweisen. Unter Zugabe von Mevalonat oder Squalen erhöht sich die Zahl kleinerer, insulinsezernierender Vesikel (Tsuchiya et al., Endocrinology 2010).

Speicherung von LDL-C in der Plasmamembran
Ein weiterer nachgewiesener Mechanismus, über den Statine in die Insulinfreisetzung eingreifen können, ist die durch Statine begünstigte Anreicherung von LDL-C in der Plasmamembran: Über die Hemmung der HMG-CoA-Reduktase senken Statine den Cholesteringehalt der Inselzelle. Als Reaktion darauf wird der LDL-Rezeptor hochreguliert und mehr LDL-C in die Zelle aufgenommen. Entscheidend ist aber nicht der absolute Gehalt an LDL-C, sondern dessen Verteilung in der Betazelle. LDL-C wird lysosomal abgebaut und gelangt in die Plasmamembran. Durch Anreicherung von LDL-C in der Plasmamembran wird Glukokinase gehemmt und damit die Insulinsekretion gestört.
Kruit et al. (Diabetologia 2010) zeigten, dass LDLR-defiziente Mäuse im Gegensatz zu hypercholesterinämischen Mäusen mit intaktem LDL-Rezeptor bei hohen Plasmacholesterinspiegeln eine normale LDL-C-Konzentration in den Inselzellen und eine normale Betazellfunktion aufwiesen. Der Mechanismus, welcher der Insulinsekretionsstörung zugrunde liegt, beruht also nicht alleine auf einer Hypercholesterinämie, sondern entscheidend auch darauf, dass LDLR in der Betazelle durch die Statintherapie hochreguliert wird.
Hemmung des Cholesterin-Efflux aus der Plasmamembran
Physiologischerweise kann Cholesterin über einen Cholesterin-Efflux-Mechanismus mit dem ABCA1-Transporter aus der Plasmamembran entfernt werden (Abb. 3). Bei Störung dieses Transporters kann LDL-C nicht aus der Plasmamembran eliminiert werden und Glukokinase bleibt inhibiert. Experimentell ließ sich zeigen, dass ABCA1-defiziente Betazellen Cholesterin akkumulieren und weitere pathologische Veränderungen wie eine veränderte Struktur des Golgi-Apparates oder verringerte Kalziumspiegel aufweisen (Kruit et al., Diabetes 2011).
Statine führen zu einer Hemmung des ABCA1-Transporters, indem der Transkriptionsfaktor SREBP2 und in weiterer Folge LDLR hochreguliert werden. Gleichzeitig wird eine in einem Intron dieses Transkriptionsfaktors befindliche mikroregulatorische microRNA (miRNA33) produziert, die wiederum den ABCA1-Transporter hemmt (Fernández-Hernando et al., Arterioscler Thromb Vasc Biol 2011). Durch Wirkung auf SREBP2 steigern Statine daher nicht nur die Aufnahme von LDL-C in die Zelle, sondern inhibieren auch die Entfernung von LDL-C aus der Plasmamembran (Brunham et al., Nat Med 2007; Sturek et al., J Clin Invest 2010). Durch Hemmung der miRNA33 mit Antagomir (anti-miR) konnte im Tierversuch die Insulinsekretion erhöht werden, dies allerdings nicht bei ABCA1-Knock-out-Mäusen (Wijesekara et al., Diabetes 2012).
Antiproliferative und antiapoptotische Effekte
Wie von Rütti et al. (Endocrinology 2009) ebenfalls gezeigt wurde, verringert LDL-C die Proliferation und Funktion, nicht aber die Apoptose muriner als auch humaner Inselzellen. Der antiproliferative Effekt der Statine erwies sich als LDL-Rezeptor-unabhängig und liefert eine mögliche Erklärung für die Resultate einer Post-hoc-Analyse, wonach Pravastatin in der WOSCOP-Studie bei Patienten mit ausgeprägter Hypercholesterinämie das Diabetesrisiko verringerte (Freeman et al., Circulation 2001). In der WOSCOP-Studie wurden Patienten mit einem mittleren LDL-C von 190 mg/dl behandelt. Möglicherweise treten bei derart hohen Ausgangswerten die insulinsekretorischen Probleme in den Hintergrund und der antiproliferative Effekt tritt zu Tage. Bei dem heute gültigen Therapieansatz „the lower, the better“ ist der antiproliferative Effekt aber wahrscheinlich weit weniger relevant als die sekretionshemmende Wirkung. Hinzu kommt, dass das in WOSCOP untersuchte Pravastatin weniger gut in die Betazellen aufgenommen wird als lipophile Statine (Ishikawa et al., J Atheroscler Thromb 2006).
In sehr hohen Mengen und möglicherweise vor allem in oxidierter Form fördert LDL-C die Betazellapoptose (Roehrich et al., J Biol Chem 2003; Abderrahmani et al., Diabetologia 2007). Eine durch Nahrungsentzug, Zytokine oder LDL-C bedingte Betazellapoptose kann durch HDL-Cholesterin (HDL-C) korrigiert werden. Das gilt auch für eine durch hohe Glukosespiegel und durch Interleukin-1β induzierte Apoptose (Rütti et al., Endocrinology 2009).
Zusammenfassung
Cholesterin ist ein wichtiger Regulator der Insulinsekretion. Zu Interferenz zwischen Cholesterinstoffwechsel, Statintherapie und Insulinsekretion gibt es mehrere Erklärungsmodelle, die durch experimentelle Daten gestützt werden. Der LDL-Rezeptor (LDLR) dürfte an diesem Effekt wesentlich beteiligt sein. Davon unabhängig wirkt sich auch der gestörte Efflux von Cholesterin aus den Betazellen negativ auf die Insulinsekretion aus. Außerdem wurde gezeigt, dass LDL-C die Betazellproliferation beeinträchtigen und in hohen Dosen bzw. in oxidierter Form die Apoptose von Betazellen induzieren kann. Im Gegensatz dazu verringert HDL-C die Apoptose von Betazellen und dürfte auch die Insulinsensitivität und -sekretion günstig beeinflussen.

Nach Vortrag von: Prof. Dr. Arnold von Eckardstein
Institut für Klinische Chemie, Universitätsspital Zürich, Schweiz

Ursprünglich erschienen:
DF 01|2013
DF 01|2013
Herausgeber: Univ.-Prof. Dr. Guntram Schernthaner, Österreichische Diabetes Gesellschaft
Publikationsdatum: 2013-03-04
Zur Ausgabe »
Publikationsdatum: 2013-03-04
Zur Ausgabe »